


FDA and regulatory flexibility
An exploration into FDA flexibility for rare diseases, setting the stage for the next 4 years

The FDA approval process often seems like a Black box, but there are patterns to the process which should allow us to develop a framework. The FDA’s trend toward flexibility for rare diseases has been in the zeitgeist, but what does “flexibility” mean? The FDA provides guidance for drug developers, but It’s instructive to analyze recent history to build a framework for future therapies. “Study the past if you would define the future.” ~Confucius.
The article goes through the history of the FDA and a few controversial decisions (Sarepta, Biogen, Amylyx, Reata) to build a framework. Part 2 of the article discusses companies with unapproved drugs where the FDA has permitted some flexibility. My framework will continue to evolve as the administration changes, but this article sets the stage for any potential change in the next four years.
TLDR:
FDA flexibility goes in cycles and we’re in a cycle of increased rare disease flexibility
P values are less important than showing signal for a drug with an unmet need
The new administration shouldn’t change much. Makary is open to new types of evidence. Vivek is a biotech guy. Marks is signaling he’d like to continue.
The FDA needs to see a dose response. The review documents and advisory committees indicate a dose response is an evidence of benefit (see aduhelm)
FDA flexibility for rare diseases hinges on unmet need. For example, the FDA has not been giving sickle cell disease where patients have some other options (see Biomarin’s issues getting a drug across the finish line).
Regulatory flexibility does not lead to commercial success. Patients and physicians have to want the therapy. (Contrast Sarepta with Aduhelm)
Tight natural history studies are ok. Some diseases have a tight natural history without variation in functional decline and the FDA is okay using that data. (See Reata)
Safety is paramount. Don’t try to approve an unsafe drug. See filgotinib, waylivra, Zuranolone
Advisory committee meetings are good to get the patient perspective to the FDA. also helpful to see how the FDA is thinking.
Introduction and History
The historically adversarial relationship between the FDA and drug developers has evolved significantly over the past decade. While the prospect of a new administration under President-elect Trump introduces some uncertainty, the underlying trend toward regulatory flexibility, particularly for rare diseases, remains robust.
The FDA's role in drug safety was established in 1938 following the deaths of over 100 people due to an impure antibiotic. This led to laws mandating pre-market safety reviews and prohibiting false advertising. In 1951, the category of "prescription-only drugs" was formalized. However, the FDA's authority remained relatively limited until the late 1950s, when the thalidomide tragedy unfolded.
“Thalidomide, widely used to treat anxiety, insomnia, and morning sickness, had not been tested in pregnant women. The US was a notable exception, thanks to FDA reviewer Frances Kelsey, who rejected the drug's marketing application, citing a need for further safety studies. Tragically, thalidomide was later found to cause severe developmental defects in fetuses. (Alex Telford).”
In response, the Kefauver-Harris Amendment of 1962 established the modern FDA, granting it the authority to approve drugs based on both safety and efficacy. This additional requirement to demonstrate efficacy became the cornerstone of the FDA's power.
Critics of the amendment argue that it led to over-regulation and a decrease in new drug approvals. Several analyses conducted between 1962 and 1985 suggested that FDA restrictions reduced the number of approved drugs while saving a marginal number of lives. Critics also point to widespread off-label drug use as evidence of excessive bureaucracy hindering access to effective treatments. (Source: dedicated FDA criticism blog).
The balance between over- and under-regulation shifted in the late 1980s during the HIV/AIDS epidemic. Facing a lack of effective treatments and certain death, activists stormed the FDA's headquarters in 1988, carrying signs with slogans like "R.I.P. killed by the FDA." This activism paved the way for the accelerated approval pathway, formally instituted in 1992. - Telford again
The accelerated approval pathway permits approval based on early-phase data if drugs demonstrate a benefit on a surrogate endpoint "reasonably likely to predict benefit in confirmatory studies." However, surrogate endpoints may not always translate to actual clinical benefits. For instance, tumor shrinkage in cancer may not always predict improved overall survival. Additionally, data from non-placebo-controlled trials often fail to hold up in larger, placebo-controlled studies.
The accelerated approval pathway permits approval based on early-phase data if drugs demonstrate a benefit on a surrogate endpoint "reasonably likely to predict benefit in confirmatory studies." However, surrogate endpoints may not always translate to actual clinical benefits. For instance, tumor shrinkage (response rate) in cancer may not always predict improved overall survival. Additionally, data from non-placebo-controlled trials often fail to hold up in larger, placebo-controlled studies.
The accelerated approval pathway has fueled criticism that the FDA is too cozy with pharmaceutical companies. Former FDA Commissioner Herbert Ley warned of "unmerciful pressure" on the agency from companies, lobbyists, and politicians. In this context, accelerated approval can be viewed as a regulatory loophole allowing the approval of marginally effective drugs. The Prescription Drug User Fee Act of 1992, which requires pharmaceutical companies to pay the FDA annually to fund operations, further strengthened these claims of bias. (Nearly a billion dollars was collected between 1993 and 2001).
However, the charges of over regulation far outweigh under regulation. Most believe the FDA is a risk averse agency who has suppressed innovation: “Over the long-term, the trend has been to steadily impose tighter and tighter constraints on drugmakers. From where we stand today, we may only be a few more tightenings of the ratchet away from a world in which the barriers to entry for new pharmaceuticals are prohibitive” - Alex Telford. But I think there’s more nuance to this statement. The FDA goes in microcycles. The thalidomide crisis precipitated a cycle of over regulation. AIDS allows the accelerated approval pathway. A tougher FDA on Oncology in the 2000s led to a swath of accelerated approvals since 2005. Criticism of the AA pathway led the FDA to create “project confirm” and attempts to regulate the pathway (analysis, patients are pissed). Peter Marks, head of the Biologics division at the FDA has pushed for increased flexibility saying: “Reviewers…. viewed themselves as “defenders of the regs and defenders of the p-value.” While regulations are important, Marks said his goal at CBER is to focus reviewers on patients, and to avoid fetishizing regulations.” - May 2024
We are now, I believe, in the early to middle stages of a cycle characterized by increased regulatory flexibility for rare diseases. While pharmaceutical companies have largely focused on large markets, such as GLP-1s, smaller companies developing therapies for rare diseases have been granted significant leeway by the FDA. Let's examine some recent examples of approved drugs and candidates in development that are benefiting from this shift.

FDA guidance Documents
The FDA publishes guidance for drug developers to guide companies on their thinking. Here are the most relevant ones in my opinion
December 2024: Accelerated Approval – Expedited Program for Serious Conditions | FDA - Most recent document with details on confirmatory trials and use of surrogate endpoints.
March 2023: Clinical Trial Considerations to Support Accelerated Approval of Oncology Therapeutics | FDA - Places more emphasis on randomized controlled trials .
Formal FDA Meetings for Sponsors or Applications of PDUFA Products Guidance on timings. Sponsors tend to meet the FDA at each stage and the FDA reports back after 1-2 months.
March 2014 Expedited Programs for Serious Conditions | Drugs and Biologics | FDA - Comprehensive criteria for every expedited program
Good article detailing Marks’ and Woodcock’s thoughts on Regulatory flexibility and the role of the FDA. May 2024
FDA guidance: Demonstrating Substantial Evidence of Effectiveness for Human Drug and Biological Products - December 2019
Drugs@FDA contains the inner workings for FDA review:
“Substantial evidence requirement for effectiveness, which had generally been interpreted as calling for two adequate and well-controlled trials, could also be met by a single trial plus confirmatory evidence. “ - The historical standard for approval.
Sarepta - Part 1 Exondys - To confirm or not to confirm? that is the question
Timeline:
March 2013 - End of phase 2 meeting to discuss potential for NDA
June 2015 - NDA submitted
April 2016 - Advisory Committee meeting (negative vote)
May 2016 - Woodcock requires longer term dystrophin skipping results
September 2016 - Unger publishes his Dissent.
December 2016 - Both Califf and Unger encourage Sarepta to republish the right results (Letter)
September 2018 - EU rejects it (negative CHMP opinion)
TBD - Confirmatory trial for eteplirsen……
Duchenne Muscular Dystrophy (DMD) is a severe genetic disorder that causes progressive muscle weakness and early death due to a lack of dystrophin, a protein essential for muscle health. Eteplirsen (Exondys 51) is a therapy developed by Sarepta Therapeutics that uses exon skipping to produce a shortened form of dystrophin, aiming to slow disease progression. Eteplirsen’s approval was highly controversial due to limited efficacy data, concerns over trial design, and intense regulatory debate.
Exondys 51 was approved because of 1 person: Janet Woodcock, Director of the Center for Drug Evaluation and Research (CDER). The major placebo-controlled trial evaluating the drug failed the primary endpoint, the FDA raised concerns over methodology and an advisory committee voted against both accelerated and full approval (7-6 and 7-3 respectively). Further the trial was in a small subset of patients (12 total) which hardly meets the “traditional” bar for a pivotal trial. But Janet Woodcock, Director of the Center for Drug Evaluation and Research (CDER), felt the drug was approvable and pushed forward.
The Advisory committee decision came in April 2016 and it was clear the FDA was leaning towards rejection. However, woodcock requested additional dystrophin data from an ongoing study to support approval saying
“If you are successful in showing, to FDA’s satisfaction, a meaningful increase in dystrophin by Western blot analysis between the paired pre-and post-treatment samples, we expect to be able to grant an accelerated approval “ review.
The results were disappointing, but Woodcock brushed it aside and recommended approval anyway. Public pressure from patients during the panel was high to approve the therapy: They wanted hope and Sarepta provided it. Dissatisfied with Woodcock's decision, Ellis Unger, the clinical reviewer, appealed to then-FDA Commissioner Robert Califf. Unger's appeal was supported by Acting Chief Scientist Luciano Borio, who expressed concerns that Sarepta "exhibited serious irresponsibility by playing a role in publishing and promoting selective data."
“Dr. Woodcock’s memorandum states that the data submitted in support of the NDA establishes “increased dystrophin protein production, a surrogate endpoint [for DMD] that [she] conclude[s] is reasonably likely to predict clinical benefit.” Dr. Unger states that he disagrees with Dr. Woodcock’s decisional memorandum because he does not believe “the findings on the dystrophin surrogate endpoint are reasonably likely to predict clinical benefit.” - Califf’s Letter
Further, Unger cited issues on how dystrophin was measured. The western blot submitted by Sarepta were “oversaturated, unreliable, and uninterpretable” requiring a re-reading by an independent pathologist. The re-read was much less favorable:
“The re-read showed a nominally statistically significant increase in dystrophin in response to eteplirsen for the low dose group, but not the high dose group [T]he type-I error rate was not controlled for multiplicity.)18 Moreover, for the 4 patients who had received placebo through Week 24 and then switched to eteplirsen, there was no increase in dystrophin at Week 48.” source
Sarepta further claimed 18% of fibers stained positive for dystrophin after 3 years, but the Western blot at the same time point indicated only 0.93% expression, a concerning discrepancy between the two measurement methods. Unger eventually raised the same concerns when the scientific article was published (link).
Bob Califf ultimately sided with Woodcock for approval and his logic is instructive. He cited genetic data on dystrophin expression and correlations between low levels and functional status to support low level dystrophin expression as a surrogate biomarker. Becker Muscular dystrophy is a well-defined disease where low levels of a truncated dystrophin lead to a less severe phenotype. Further, he emphasized that the significant unmet need in DMD outweighed any shortcomings in the development program. Califf stated that this flexibility was justified due to several specific factors, including: the life-threatening nature of the disease, the lack of available therapies, the intended population is a small subset of an already rare disease, and DMD is a fatal childhood disease. Of note, the therapy has been “relatively safe in the clinic”
He agrees lowering the bar for approval may expose patients to undue harm but is confident “this unique situation will not set a general precedent”. Further he defers to Woodcock seemingly because she has the default authority even though he shares similar concerns on the evidence and processes from Sarepta. Califf further emphasizes “ it is critical that….[Sarepta]... must conduct the required confirmatory trial with due diligence.
Califf may want a mulligan on this one.
Sarepta’s Exondys is still under accelerated approval 8 years later. The confirmatory trial is set to complete later this year and one can hope we have some data on functional outcomes. The original deadline was 2020 which has come and gone. Sarepta mentions “accelerated approval” as a risk in their SEC filings but feels no need to mention timelines for confirmatory studies. They cite growing revenues and answer commercial questions instead. They’ve made billions off of the therapy. Without the accelerated approval, Sarepta would have likely gone bankrupt.
Sarepta - Part 2 Elevidys - Peter Marks’ FDA
Timeline
May 2023 - Advisory Committee votes 8-6 in favor of accelerated approval.
June 2023 - FDA recommends against approval but Peter Marks overrules them and grants AA for boys aged 4-5
October 2023 - EMBARK confirmatory trial reads out, Misses primary endpoint, favorable trends.
June 2024: marks over rules FDA staff again to grant full approval for ambulatory boys older than 4 and AA for non ambulatory boys.
The Sarepta Saga continues with their recently approved Gene Therapy for Duchenne Muscular Dystrophy: Elevidys. Duchenne Muscular Dystrophy is caused by a lack of functional dystrophin protein, and a gene therapy tries to replace it with functional protein. However, since the gene is so large, it can only replace dystrophin with a shortened ”microdystrophin”. The correlation between microdystrophin expression and functional clinical benefit remains unclear, because it’s a novel creation from the gene therapy. This uncertainty is further compounded by Pfizer's recent failure with its DMD gene therapy, where dystrophin expression did not translate into improved functional outcomes. (webinar)
May 12, 2023: An advisory committee convened to discuss Sarepta's Elevidys and voted in favor 8-6 for accelerated approval. Sarepta presented results from 2 main studies.
Study 102: A placebo-controlled study involving 40 patients. The primary endpoint was not statistically significant, but a subgroup analysis suggested potential benefit in younger patients.
Study 103: A study involving 20 patients, using matched external controls.


Elevidys demonstrated a potential benefit on the North Star Ambulatory Assessment (NSAA) in a small subset of patients (8 patients) aged 4-5 years. However, no clear association was observed in older patients. Post-hoc analyses suggested a potential benefit in a subgroup of ambulatory patients aged 4-5 but not in those aged 6-7. It is also worth noting that the group receiving Elevidys did not show overall improvement.
Notably, the confirmatory study was fully enrolled (unlike the eteplirsen case) and set to readout soon. The advisory committee voted 8-6 in favor of approval, largely because the drug was safe and “time is muscle” (source)
However, the FDA disagreed with the Adcom and said
“The data in your BLA do not indicate a persuasive correlation between expression of ELEVIDYS micro-dystrophin and improvement on the North Star Ambulatory Assessment. Thus, there is insufficient evidence that expression of ELEVIDYS micro-dystrophin is “reasonably likely to predict clinical benefit” to support Accelerated Approval of ELEVIDYS.” (source)
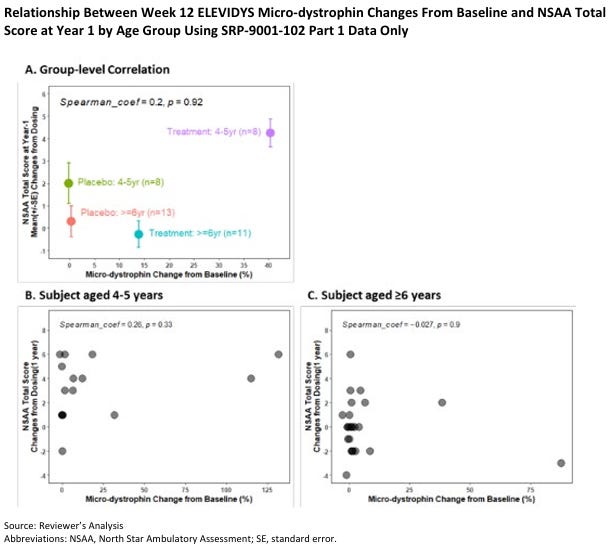
Elvidys was approved in the summer of 2023.
What?
Yes. Peter Marks, in a historic decision, reversed the FDA decision and granted accelerated approval.
“I disagree with certain interpretations of the efficacy data and come to a different conclusion regarding individuals ages 4 through 5 years. “
People can disagree with his decision, but the full study results would be available in the fall of 2023 and the drug would be withdrawn if it failed
In October of 2023, the confirmatory study failed on the primary endpoint but showed some positive trends on secondary endpoints. There was a benefit on the time to rise and 10 meter walk test but the NSAA (primary endpoint) did not hit statistical significance). But Sarepta was optimistic citing “Results support submission of an efficacy supplement to the BLA; US FDA has indicated openness to reviewing the data for label expansion based on the totality of evidence from EMBARK”
Sarepta’s stock was down 30% on the day but quickly recovered as investors likely realized the flexibility afforded to Sarepta would lead to approval. Some of the key secondary functional endpoints did hit statistical significance so the case could be made for approval. However, the FDA ultimately decided against conversion to full approval saying:
“Specifically, it is my assessment that the data submitted in the sBLA: • do not verify the clinical benefit of Elevidys in ambulatory boys aged 4-5 years (i.e., the group for which accelerated approval was granted by Center Director override of the review team and senior CBER leadership recommendation), • do not demonstrate the benefit of Elevidys in ambulatory patients greater than age 5 years of age, or • do not demonstrate the benefit of Elevidys in non-ambulatory patients of any age.” (source)
Elevidys was granted full approval for boys aged 4-5 in 2024. Peter Marks over ruled the FDA again

One thing is for certain: Peter Marks wanted those drugs approved. And as we’ll see, the increased regulatory flexibility extends to other areas as well.
Marks Letter 2: https://www.fda.gov/media/179484/download June 24
Marks Letter 1: https://www.fda.gov/media/169715/download June 23
Duchene approval exposes FDA rift over Sarepta gene therapy | BioPharma Dive
3. Biogen - Aduhelm - Approved but no one used it
March 2019 - Discontinuation for futility
October 2019 - Announce plans to pursue approval based on seeing a dose response trend.
July 2020 - BLA submission
November 2020 - 10-1 vote against in an AdCom (note the FDA is still positive)
June 2021: AA granted based on Amyloid beta plaque reduction
July 2021: Resignations and amendment to approval to narrow population
April 2022: Medicare covers the drug only for clinical trial patients.
Jan 2024: Discontinuation of commercial efforts.
Aduhelm's accelerated approval is one of three recent neurology drugs approved under controversial circumstances. Biogen originally discontinued two large phase 3 trials for futility in March 2019 but reversed course in October that same year after analyzing a high dose cohort from one of their trials. Essentially, Biogen saw a dose response and the trials failed because of issues with the trial protocol. As usual with controversial applications, an advisory committee convened in November 2020. In contrast to the Sarepta case, many FDA staff members were in favor of approval, while the advisory committee nearly unanimously voted against the drug (10-1). Similar to the Sarepta situation, many patients and advocacy groups supported approval, with some even comparing aducanumab to AZT, the first HIV/AIDS drug.
The advisory committee itself is a fascinating case study in how investors “read tea leaves”. Before any advisory committee meeting, briefing materials are made public. The FDA meeting materials were more positive than expected and Biogen’s stock rose 40% (source). Once the advisory committee convened and voted 10-1 against approval, the stock whipsawed back down, all within the week.
Despite the advisory committee's strong opposition, the FDA staff remained enthusiastic, and internal advocates continued to push for approval. The FDA (and Biogen) used a post hoc subgroup analysis to make the case for a dose-exposure response relationship. A protocol amendment during the trial led to higher doses given to certain participants and the trial which demonstrated a benefit had more patients exposed to the higher dose.
Doctors were not convinced because only 1 of 4 subgroups demonstrated a benefit on the primary endpoint (Table)
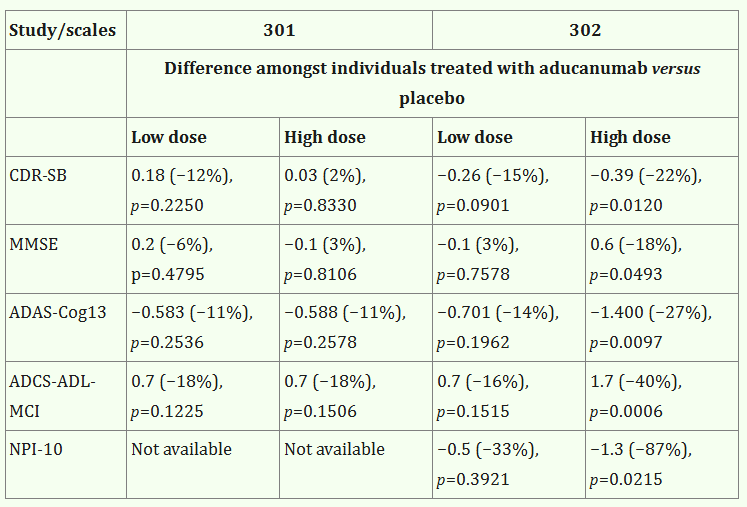
Aducanumab was granted accelerated approval in June 2021, based on its impact on the surrogate biomarker amyloid-beta plaque, as measured by amyloid PET (accelerated approval was not discussed at the panel). The FDA concluded that: 1) amyloid plaque is clearly associated with Alzheimer's disease, 2) group-level relationships demonstrate dose-dependent decreases in amyloid levels (somewhat supported by data), and 3) the relationship between amyloid reduction and functional outcomes is established (more debatable). The high unmet need combined with some effect on a surrogate biomarker supported approval
“ Even given the residual uncertainty with respect to its clinical benefits, patients and caregivers have clearly stated their desire for a drug that is likely to be effective. This is exactly the situation for which accelerated approval exists – where the evidentiary criteria for accelerated approval are met, it can provide earlier access to a promising drug to patients with unmet needs. “
Subsequently, several "powerless" advisors resigned, stating the FDA had made a "mockery" of the adcom’s role by disregarding their recommendation.
Lots of ink has been spilled on the topic so I won’t rehash it (see a quick perplexity search) but a few things stand out.
The approval is unique among the rest because Alzheimer's is a highly prevalent disease, and the FDA disagreed with the advisory committee (unusual).
Billy Dunn seems like a key player advocating for approval and the internal decision prompted a house of representative investigation.
The FDA latched on to the biomarker data, dose response relationship, and heavy patient advocacy to make their decision.
Aduhelm ended up a failure unlike Sarepta. Biogen wanted to charge 56,000 per year for the drug so people were rightfully up in arms citing potentially 29$ billion dollars in potential revenue for a drug that doesn’t work. Medicare disagreed.
CMS restricted coverage for Aduhelm to patients in clinical trials and after a brief clinical struggle, the drug was pulled in November 2024.
Three F.D.A. Advisers Resign Over Approval of Alzheimer's Drug - The New York Times
4. Skyclarys - Skyclarys (Omaveloxolone) and Bardoxolone
Reata Pharmaceuticals provides another interesting case study, involving two drugs: bardoxolone for Alport syndrome and Skyclarys (omaveloxolone) for Friedreich's ataxia. While Skyclarys' approval journey is particularly noteworthy, the experience with bardoxolone offers valuable context.
Bardoxolone was a repurposed drug for Alport syndrome, a form of kidney disease. Alport syndrome is another hereditary, progressive disease with high unmet need and few available treatment options. Before being tested in trials for Alport syndrome, Bardoxolone was studied in diabetes patients with chronic kidney disease (CKD) in the BEACON trial. However, that trial was terminated early due to safety concerns, which later carried over to its trial in Alport syndrome.
While Bardoxolone demonstrated a temporary increase in glomerular filtration rate (GFR), the number of patients progressing to kidney failure (the end goal is reducing kidney failure rate, not GFR!) was the same between treatment and placebo arms. Although GFR is often used as a marker for kidney function, an increase in GFR does not necessarily indicate better kidney function. In fact, temporary increases can overwork the kidneys, potentially leading to worse long-term outcomes.
Moreover, several patients discontinued the drug due to liver toxicity, and nearly 90% experienced some form of liver enzyme elevation—a concerning indicator of potential liver damage. These safety concerns, coupled with the lack of long-term benefit, led the FDA Advisory Committee to unanimously vote against approval in late 2021. A Complete Response Letter (CRL) was issued in February 2022.
Skyclarys
Skyclarys' approval was based on a single, randomized, double-blind, placebo-controlled study (MOXIe Part 2), its open-label extension study (MOXIe OLE), confirmatory evidence from a natural history comparison, and pharmacodynamic data supporting the proposed mechanism of action. Let’s walk through a timeline
October 2019: initial Data release from MOXIE
November 2020: FDA says trial is not enough
Jan 2021: plans for a second pivotal study.
Mid-2021: FDA Changes Course
First quarter of 2022: Reata submits and NDA is accepted by the FDA. Adcome scheduled
Mid 2022: Reata submits additional OLE analysis. FDA pushes back PDUFA to February2023
October 2022: Adcom cancelled FDA scraps AdComm for Reata’s rare disease candidate
November 2022: Reata explains their strategy and timeline. After a recent late cycle meeting. The FDA confirmed they were looking at data.
Feb 2023: approval
All FDA review documents found here: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/216718Orig1s000TOC.cfm
October 2019: Initial Data Release
Reata released the pivotal MOXIe trial results for Friedreich's Ataxia, demonstrating a statistically significant improvement in the mFARS (modified Friedreich’s Ataxia Rating Scale) primary endpoint.
November 2020: FDA Skepticism
Despite positive trial data, the FDA expressed doubts:
"It does not have any concerns with the reliability of the modified Friedreich’s Ataxia Rating Scale (“mFARS”) primary endpoint results from the registrational Part 2 of the MOXIe trial (“Part 2”) of omaveloxolone in FA patients. However, it was not convinced that the results from Part 2 support a single study approval."
Reata proposed using baseline-controlled data as an alternative to a second randomized trial. This approach compared patient-level natural history data to measure progression pre- and post-treatment. Both the MOXIe trial and baseline-controlled analysis met the primary endpoint.
January 2021: Plans for a Second Pivotal Study
Despite ongoing discussions with the FDA, Reata anticipated the need for a second pivotal study:
"We plan to initiate a second pivotal study in the second half of 2021, following discussions with the FDA and the European Medicines Agency (“EMA”)."
Mid-2021: FDA Changes Course
The FDA signaled a shift by scheduling a pre-NDA meeting, suggesting the existing data package could support an NDA submission. Reata clarified:
"So for the pre-NDA meeting, it's primarily -- we may discuss the mechanics of submission of the NDA. And so we do not anticipate there to be any meaningful discussion of interpretation of clinical efficacy or safety data. We commented before that when they asked us to withdraw from the Type C meeting and submit for the request for the pre-NDA meeting, they told us to redirect our briefing materials to the topics that would typically be covered in the pre-NDA meeting.
for us to determine if there will be any additional trials required in the post-marketing setting. FDA had previously signaled that we needed additional evidence to support an NDA submission for marketing authorization. And so we believe that the data that we provided them in the baseline controlled study as well as the other data sets have allowed us to overcome that hurdle. And so we do not believe at this point that there will be another premarketing trial needed, but we'll determine at the meeting if a post-marketing trial may be needed." - Reata August 2021
The FDA documents are notably more even keeled (from administrative correspondence: “The exploratory data analysis does not intend to imply a degree of endorsement for efficacy.” - FDA August 2021
First quarter of 2022: Reata submits and NDA is accepted by the FDA.
November 2022: Reata explains their strategy and timeline.
Note this was after the advisory committee meeting was cancelled: Link.
“In the third quarter of this year, we completed a mid-cycle communication meeting with the FDA and submitted additional data and analysis to the FDA in response to their comments.
The FDA determined that these submissions were a major amendment to our NDA and extended the Prescription Drug User Fee Act or PDUFA date by 3 months to provide time for a full review of the new data and analyses. The PDUFA date is now February 28, 2023.
Next slide. We recently completed a late-cycle meeting with the FDA. The purpose of the late-cycle meeting is for the FDA to discuss any substantive issues identified in the division's objectives for the remainder of the review. The meeting does not address the final regulatory decision for the NDA. While we have not received formal minutes from the FDA, in the preliminary agenda 4 and during the late-cycle meeting, the FDA stated that they continue to review the analyses and data included in our recent NDA submissions.
The FDA did not request any additional data or analyses but stated that additional data may be requested as reviews are ongoing. The FDA confirmed that no information requests were outstanding and reiterated that they do not currently plan to hold an advisory committee meeting.”- November 2022.
The Advisory Committee cancellation had many speculating on reasons, considerations, and chances for success. I believe the Adcom was cancelled after Reata submitted additional OLE data supporting approval.
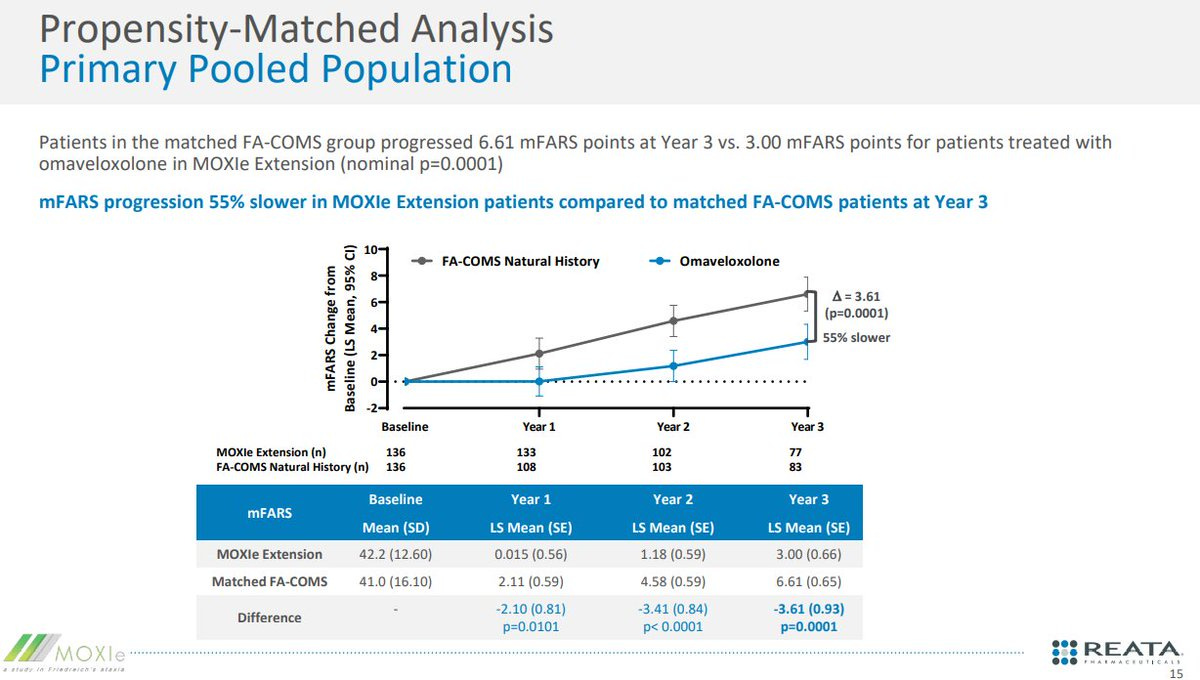
The FDA's primary concerns about the Skyclarys application were consistent throughout the review process: the small size of the pivotal trial, the reliance on open-label extension data, and debates surrounding the utility of the withdrawal design (where the drug is stopped to observe the patient's response). The FDA noted these concerns and still approved the drug because the propensity matched analysis (match patients to a similar population) showed a consistent benefit.
“The chosen matching factors appear to be clinically meaningful covariates that were appropriate for inclusion.” The reviewer mentions the high 30-50% attrition rate, post hoc nature, limitation on specific covariates (GAA repeats), non matching durations, but the conclusion is that the natural history provides confirmatory benefit because the natural history is tight. Magnitude of deterioration in mFARS is the same regardless of the number of patient’s " - FDA review.
The unmet need and patient advocacy certainly played a role in approval: 74,000 people signed a petition for approval in 2021 (link)

See how the stock reacts. October 2019 data release and it’s up 100%. FDA requests additional trials and the stock reacts poorly. When the “pre-NDA meeting" is announced in 2021, the stock is up. But after the failure in Alport, no one believes in the approval until it happens in early 2023.
Amylyx and Relyvrio
Once again, let’s walk through the data and a timeline.

Courtesy of Parexel
Paraxel has compiled an amazing timeline and story for the drug so I’ll direct you to their article. Their key takeaways were
For now, FDA has shown flexibility in considering approval for drugs for diseases with limited treatment options and significant unmet needs, particularly neurodegenerative diseases. However, this flexibility may wane if scrutiny of therapies approved with modest data packages continues to mount.
Patient advocacy groups are playing an increasingly important role in facilitating development and approval and drug makers should nurture relationships with them.
Pricing is coming under increasing scrutiny and companies need to be prepared to provide a robust defense of their pricing.
I also have a few key takeaways.
The first advisory committee meeting was negative because of “small sample size, missing data, and baseline imbalances”. The committee simply did not believe the Centaur Trial was significant. Note the primary endpoint was positive using the prespecified analysis but a separate FDA analysis moved the p value above 0.05. Stats ruled the world
Between meeting one and two, additional analysis conducted demonstrated a survival benefit for Relyvrio. The new data shows a dose response and benefits on time to first hospitalization. No new trials were run but new analysis and longer term follow-up were positive.
The new panel expressed the same concerns but were swayed by the panel and Billy Dunn’s involvement. Billy Dunn made the company CEO promise to withdraw the drug if the confirmatory trial failed. This actually sways a couple people to vote in favor of approval. The drug was granted full approval because the endpoints in the trials were functional endpoints, not surrogate.
Eventually, a phase 3 trial (PHOENIX) designed to confirm the benefit for Relyvrio failed in March 2024. However, Amylyx still made nearly 400$M in 2023 from the drug. Patients believed the drug works and neurologists were okay prescribing it. Contrast the situation to Aduhelm where access was low and no one prescribe the drug.
Some examples where the Same flexibility wasn’t applied
Sage therapeutics in Major depressive disorder and postpartum depression - Zuranalone was only approved in postpartum depression. The FDA had concerns on the efficacy and safety evidenced by the Black box warning for drowsiness. Still approved for PPD, higher unmet need.
Biomarin - Roctavian for Sickle Cell Disease - The FDA required additional safety and efficacy data in 2020. One year of follow-up was not enough (notably different from some of the other therapies we’ll discuss). Sickle Cell Disease has many more treatment options
Rejection of Waylivra - Waylivra is an antisense Oligonucleotide therapy for Familial Chylomicronemia Syndrome, a rare disease. An Advisory Committee voted in favor of approval but the FDA had concerns over safety and late amendments to the application. The EU did approve the therapy (link)
Applied therapeutics for Galactosemia (2024) - This case is unique because the data was truly horrendous. Further, the FDA has major concerns about the trial conduct.
My major takeaways
The FDA needs to see a dose response. The review documents and advisory committees indicate a dose response is an evidence of benefit (see aduhelm)
FDA flexibility for rare diseases hinges on unmet need. For example, the FDA has not been giving sickle cell disease where patients have some other options (see Biomarin’s issues getting a drug across the finish line).
Regulatory flexibility does not lead to commercial success. Patients and physicians have to want the therapy. (Contrast Sarepta with Aduhelm)
Tight natural history studies are ok. Some diseases have a tight natural history without variation in functional decline and the FDA is okay using that data. (See Reata)
Safety is paramount. Don’t try to approve an unsafe drug. See filgotinib, waylivra, Zuranolone
Certain divisions (neuro) are certainly harder on drug developers but the trend is away from that. Key players in the organization can drive the culture and make decisions (Dunn, Woodcock, Marks, etc). Decisions can come down to a few select individuals

The Future:
I believe the trend towards increased flexibility will continue. Vivek Ramaswamy, head of DOGE (Department of Governmental Efficiency) has signaled the FDA is too strict on drug developers and wants to accelerate the process. Marty Makary is open to novel trial designs and new therapies (microbiome, psychedelics, etc). Peter Marks has signaled his ongoing support for rare disease flexibility.
Regulations will continue to be strict for large populations. The unmet need has to be real for FDA flexibility.
Confirmatory trials will be enforced. I anticipate the FDA will make shifts toward enforcement on confirmatory trials. Sarepta has made a joke of this process, but the FDA won’t allow that to occur again.
Supplement: Drugs in development with Flexibility included.
Above is a (non-comprehensive) list of companies who have the blessing of the FDA for some flexibility or are potential candidates. I have brief notes on a few
Stealth elamipretide for Barth syndrome - Still in progress
2021: FDA files a refusal to file letter for the NDA from Stealth stating the evidence was not enough.
Late 2021: Stealth still plants to submit the NDA
June 2022: new OLE data leads to preNDA meeting and plans to file (source)
Early 2024: Stealth BioTherapeutics submitted a New Drug Application (NDA) for elamipretide to treat Barth syndrome.
April 3, 2024: The FDA agreed to file and review the NDA for elamipretide7.
April 8, 2024: The FDA officially accepted the NDA for filing
September 9, 2024: Stealth announced that the FDA would hold an advisory committee meeting on October 10, 20242.
October 10, 2024: The FDA's Cardiovascular and Renal Drugs Advisory Committee (CRDAC) met to evaluate the NDA for elamipretide
Despite the FDA's initial skepticism, the committee voted 10-6 in favor of recommending approval. The controversy stemmed from elamipretide's failure to meet its primary endpoints in the TAZPOWER trial. However, the committee's support was largely influenced by the lack of alternative treatments and the challenges of conducting trials for ultra-rare diseases. As one committee member noted, "We're dealing with an impossible situation here. The data aren't perfect, but the unmet need is undeniable." This sentiment echoes the growing call for regulatory flexibility in evaluating treatments for rare diseases, where traditional clinical trial designs may not always be feasible or appropriate
Rocket Pharma - Danon Disease Gene Therapy
Rocket Pharmaceuticals is developing RP-A501, a gene therapy for Danon disease, a rare and fatal genetic heart condition primarily affecting young boys. Danon disease is characterized by an enlarged heart due to a deficiency in the LAMP2 protein, crucial for clearing cellular debris. Without a heart transplant, boys with Danon disease typically do not survive past the age of 20. There are currently no approved therapies.
RP-A501 aims to restore LAMP2B protein expression. The development path hasn't been without its hurdles; the FDA initially placed a clinical hold on the treatment out of an abundance of caution (link). However, after the Phase 1 trial began, the FDA reversed course, granting Rocket significant leeway regarding the endpoints for accelerated approval. The primary endpoints are now LAMP2 expression and a 10% reduction in left ventricular mass index (LVMI) at 12 months. The traditional standard requires functional endpoints and long term followup, but the FDA has weighed, risks/benefits and granted flexibility. A recent publication in the New England Journal of Medicine and accompanying investor presentation, indicates that every patient in the trial met this predefined threshold for approval.
I believe RP-A501 will be approved, and furthermore, that the prevalence of Danon disease is higher than currently estimated by investors. Rocket will host an epidemiology day in the first half of 2025, which should provide more clarity on the market size. This case exemplifies the FDA's willingness to accept biomarker data as a basis for accelerated approval in ultra-rare, life-threatening diseases with no other treatment options.
Capricor - CAP-1002 for Duchenne Cardiomyopathy
I’ve written about Capricor before in detail. Capricor's journey with CAP-1002 for Duchenne cardiomyopathy is particularly fascinating because it was the FDA chnaged course after seeing already known data.
To recap, Capricor faced challenges enrolling their HOPE-2 trial during the COVID-19 pandemic. This trial focused on Duchenne-related cardiomyopathy, a serious manifestation of DMD. The trial ultimately failed to meet its primary endpoint but showed some positive trends in secondary endpoints. Subsequent open-label extension data demonstrated a potential benefit in less than 10 total patients.
But the FDA was okay with them submitting for approval “as part of our pre-BLA meeting in August, when we show them the cardiac data from the HOPE-2 open-label extension study compared to the natural history data set, it became clear that deramiocel was slowing the trajectory of cardiac dysfunction in DMD measured by ejection fraction and measurements of end systolic and end diastolic index volumes. FDA noted the strength of our data and also noted that there were no approved therapies for cardiomyopathy associated with DMD. Based on that meeting and subsequent meetings with FDA, we have decided to move forward to file for full approval”
Keep in mind they plan to file for full approval and then expand the market. They previously planned to report HOPE3 results (phase 3 trial) later this year, but are now planning to file for approval using HOPE 2 and delaying the HOPE3 readout (why risk it?)
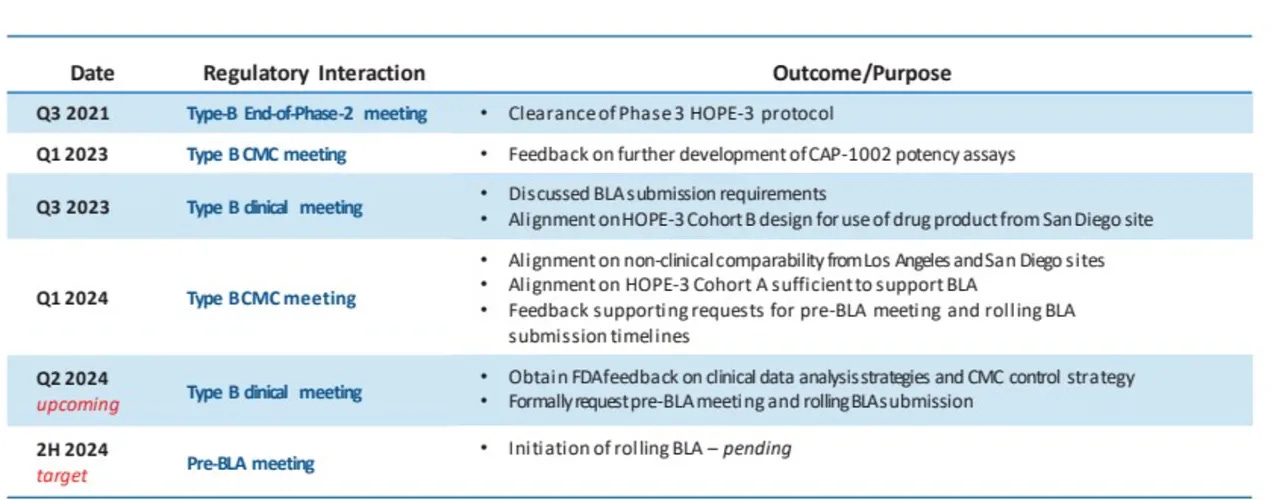
This case highlights the FDA's willingness to consider open-label extension data, even from a small patient population, when the unmet need is high
Soleno Therapeutics - DCCR for Prader-Willi Syndrome:
Soleno's experience with DCCR for Prader-Willi Syndrome (PWS) mirrors the pattern we observed with Reata's Skyclarys: a single, somewhat controversial study followed by open-label extension data that bolsters the case for approval. The timeline for DCCR is strikingly similar:
January 2020: Enrollment completed for Phase 3 DESTINY PWS (C601) study.
June 2020: Top-line results from DESTINY PWS announced.
February 2021: Pre-COVID data analysis from C601 shows statistical significance in efficacy endpoints.
September 2021: Interim one-year data from C602 (OLE study) shows significant reductions in hyperphagia.
January 2022: FDA Type C meeting recommends additional controlled data for NDA submission.
March 2022: Amended protocol submitted for Study C602 to include a randomized withdrawal (RW) period.
October 2022: Initiation of RW period for Study C602.
May 2023: RW period enrollment completed.
September 26, 2023: Positive top-line results from the RW period announced.
April 26, 2024: Peer-reviewed article published in the Journal of Neurodevelopmental Disorders.
April 29, 2024: Granted Breakthrough Therapy Designation by the FDA.
June 1–4, 2024: Data from C602 RW period presented at ENDO 2024.
June 28, 2024: NDA submitted to the FDA.
August 7, 2024: FDA grants Priority Review with a PDUFA target action date of December 27, 2024.
November 28, 2024: PDUFA date extended.
Just like with Skyclarys, the FDA initially requested additional data but eventually accepted the NDA. The recent PDUFA extension raises the question: what constitutes the "major amendment"? It's plausible that the publication of the full data set in a peer-reviewed journal triggered this extension. A cursory review suggests the data are positive and could support approval.
One notable difference is the level of interaction with the FDA. Soleno filed after receiving feedback but without extensive back-and-forth meetings, unlike Reata. This suggests that the FDA may be comfortable with a less intensive review process when the data, particularly from open-label extensions, are compelling and the unmet need is clear. I lean toward approval: the FDA
Lexeo Therapeutics - LX1001 for Friedreich's Ataxia Cardiomyopathy:
The FDA extended the same flexibility from Rocket Pharmaceuticals to Lexeo. Lexeo is running early trials for a gene therapy in Friedreich's Ataxia (FA) Cardiomyopathy. FA is a neurological disease, but cardiomyopathy is the primary cause for death in patients. The cardiomyopathy is a slowly I wrote "Management guided to the following targets as goals for accelerated approval with the FDA: 5% frataxin expression, 40-50% IHC, 10% reduction in LVMI, and 30% improvement in troponin. IHC measures % of cells expressing the protein, Frataxin level measures % of normal total protein. ".
The FDA has agreed to consider an increase in frataxin expression and a reduction in LVMI as co-primary endpoints for accelerated approval, with target levels of a 10% reduction in LVMI and 40% frataxin-positive area as measured by IHC. They also support enrolling patients with elevated LVMI. Although there is currently no direct evidence demonstrating that increased frataxin expression leads to clinical benefit in FA cardiomyopathy, the FDA is willing to consider accelerated approval based on single-arm data. Deadly disease + Unmet Need + CBER = flexibility.
Edgewise Therapeutics - Sevasemtan for Becker Muscular Dystrophy:
Edgewise Therapeutics is a dark horse in this race, with its recent positive topline results for Sevasemtan in Becker Muscular Dystrophy (BMD). BMD is a milder form of DMD, where the dystrophin protein is shorter but still partially functional. Patients experience a gradual decline in muscle function, typically becoming severe between the ages of 20-40. There are currently no approved therapies for BMD.
Edgewise reported positive results from its CANYON trial, demonstrating improvements in both the primary endpoint, creatine kinase (CK, a marker of muscle damage), and the North Star Ambulatory Assessment (NSAA), a functional endpoint. The FDA has been skeptical of CK as a primary endpoint in muscular dystrophies (because CK levels can be confounded by muscle loss over time), but the NSAA is a well-established endpoint that has been used to support approvals in DMD (see Sarepta Section).
I suspect Edgewise will approach the FDA to discuss the possibility of accelerated approval. Given the positive NSAA data, the lack of approved therapies for BMD, and the precedent set by approvals in other muscular dystrophies, I believe they will be met favorably.

Regulus Therapeutics - Cyst Volume not enough for thee but enough for me
Regulus is interesting because the regulatory path represents a clear shift from 15 years ago. In the 2000s, the FDA scoffed at using as a total kidney volume as a surrogate marker for approval in patients with Autosomal dominant polycystic kidney disease. They required Otsuka to run a multi-year study to show functional benefit for Tolvaptan (now approved). This was the FDA’s perspective on Tolvaptan:
“the Agency has consistently denied effects on kidney volume as an adequate surrogate for progressive loss of renal function” - FDA review document
Contrast that perspective with now:
“The FDA designated TKV as a reasonably likely surrogate marker for disease progression in ADPKD, which could serve as an endpoint under an accelerated approval pathway” - 2023 summit
Regulus has said the FDA is open to using Cyst Volume as a surrogate endpoint they will be meeting the FDA in the fourth quarter of this year.
Proposed model of sentiment

Conclusions
There’s a lot of flexibility going around for rare diseases. I expect it to continue. Bigger indications will face some scrutiny, but the FDA is open to novel trial designs. Today’s FDA is not yesterday’s FDA. “Don’t fight the Fed FDA”
Let me know your thoughts.
Adu Subramanian
As a further example of regulatory flexibility, consider Soliris (Alexion) for Myasthenia Gravis. It failed P3 (p>.05) but was approved anyway based on totality of evidence. In those days, there were relatively few treatments for MG- times have changed but it underscored your point.