
FDA flexibility with Capricor
FDA wants your drug to be approved. Let's look at Capricor, Lexeo, and Rocket

"I come to a different conclusion regarding the overall interpretation of the data" - Peter marks. Center Director Decisional Memo - ELEVIDYS (fda.gov). Marks’ FDA is not your grandpa’s FDA
Agenda: Looking at a few companies and trends at the FDA
DMD history at the FDA - “The betamax of gene therapy”
Capricor’s History - FDA does it again
Friedrich’s ataxia and Lexeo - Can history repeat?
Danon and Rocket Pharmaceuticals - Low bar for approval, big market, unmet need
"The Beta max of Gene therapy": FDA loves DMD drug developers.
The arc of the FDA bends toward stringency (some think Tylenol wouldn't be approved under today's standards link) but the advent of the accelerated approval pathway and increasing emphasis on patient needs have led to some notable approvals in recent years. Drugs like Exondys, Elevydis, Aduhelm, Skyclarys, and Relivryio are among a slew of drugs approved where patients’ need triumphed.
Advocates for flexibility argue that unmet need triumphs strong statistical evidence. But some patients are angry over how some companies take advantage of FDA leniency, specifically in Duchenne Muscular Dystrophy, to make billions.Stunning patient rant. That patient was tired of empty promises from Sarepta and tore them a new one. **Sarepta then tried to hide it.
From the earlier patient - "You don't do any of the follow up studies. you don't produce any of the data. if you're looking for your son who is not ambulatory to get the results of some study, fat chance....you're the beta max of gene therapy" (patients would be excluded from other gene therapies).
I believe the Accelerated approval pathway is positive but can be perverted. I'd rather err on the side of approving ‘iffy’ drugs and leaving judgment to patients and doctors. But the FDA has to enforce the rules. Sarepta used surrogate markers to gain approval for most of their drugs and the FDA does not enforce requirements for confirmatory studies. EXONDYS 51 was approved in 2016…..and remains under accelerated approval. You can see the other drugs still approved under accelerated approval here.

Sarepta’s newest gene therapy, Elevydis exemplifies FDA leniency in Duchenne Muscular Dystrophy. The drug was approved under accelerated approval (controversial), subsequently failed the confirmatory study, but was still granted full approval by an overriding decision from Peter Marks, a Division head.
“I come to a different conclusion regarding the overall interpretation of the data" - Peter marks. Center Director Decisional Memo - ELEVIDYS (fda.gov). He reversed the opinion of others in the FDA.
Mark's FDA is not your grandpa’s FDA.
Capricor: The FDA blesses another company.
The FDA gave hope to Capricor Therapeutics after the company was left for dead. Keen observers could have predicted this from their Q2 conference call.
CAP-1002 - Cardiac Stem Stem Therapy
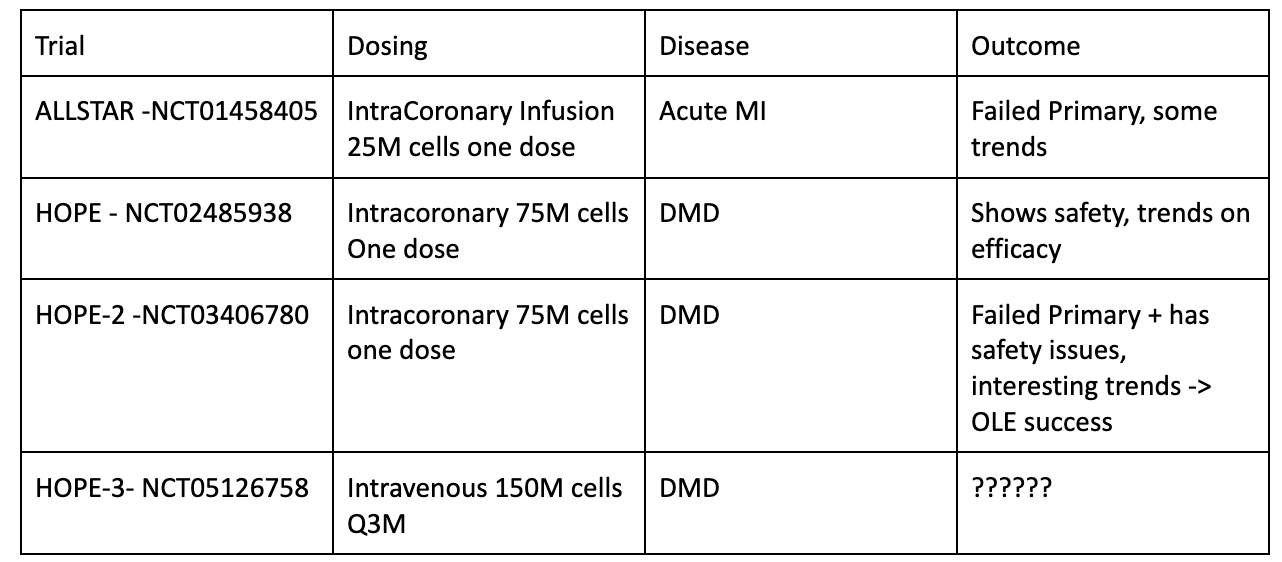
CAP-1002 has failed more trials than it has suceeded. Capricor uplisted to Nasdaq in 2015 trying to treat three diseases with CAP-1002: myocardial infarction, heart failure and DMD related Cardiomyopathy. All three trials didn't work out. The drug consistently showed some activity but never enough to run a phase 3 trial or file an application with the FDA. Despite the marginal results, the company maintained a sense of optimism common in small cap biotech. Their large pharma partner (Janssen) leaves them in 2017, but the company keeps on chugging (link).
The HOPE trial (single administration of CAP-1002) treating DMD - Cardiomyopathy (a severe and often deadly manifestation of DMD) shows the strongest signal so they start another phase 2 trial, HOPE-2, testing multiple doses in DMD patients with advanced disease. HOPE 2 planned to enroll 80 patients (spoiler: they didn't get there) with registrational intent using a difference on the mid PUL score at month 12 (measures muscle function). However, during enrollment, The company paused HOPE 2 due to a severe anaphylactic reaction in one of the patients. The company "capped enrollment at 20 patients after an interim analysis showing clinical relevance" (Sep 2021, This is what the kids call 'Gaslighting', the trial likely stopped because of safety issues and long delays after the hold). The FDA supported the PUL 2.0 mid-level test but did not explicitly clear the trial to serve as a registrational trial.
"The FDA advised Capricor to request an end of phase meeting after completion of the trial to determine whether HOPE-2 could serve as the registration study….. In addition, the agency stated that the trial would need to provide evidence of clinically meaningful changes in the PUL, as well as other evidence supportive of CAP-1002 efficacy for patients with advanced Duchenne muscular dystrophy, in order to serve as a registration trial." - January 2019
FDA support was clearly wavering
HOPE-2 reads out in May 2020. The trial misses on Mid-level PUL 2.0 but hits on the original endpoint Mid-level 1.2. The trend is positive and favors CAP-1002 on both functional and cardiovascular parameters.
Since the trial was powered for 80 patients, stats situation is funky:
"Once they adjusted the model for sample size and performed the appropriate statistical analysis, they determined that the significance of the data went from p equals 0.08 to p equals 0.01" - translation: we need some funky stats to hit the primary.
I'll note the safety isn't perfect either: 3/8 patients had a hypersensitivity reaction to the product and the trial was placed on hold due to a severe anaphylactic reaction.

The FDA ultimately requires them to run a phase 3 registrational trial to get the drug approved.
"they[FDA] felt that the sample size is too small to grant accelerated approval and so requested that we conduct an adequate and well-controlled Phase III clinical trial" - Management in September 2021.
Between 2022 and 2023, The timeline is as follows: Í

Regulatory Timeline

Then it get's interesting in the 2Q earnings call.
The plan was to report Cohort A in 4Q24 and initiate a BLA. The plan was to use Hope 3 as the registrational trial. The plan went out the window. And management couldn’t hold their excitement on the Q2 call
"I can share that we are discussing alternative paths to our BLA filing. One pathway would be filing our BLA with currently available data from the HOPE-2 and the HOPE-2 OLE studies to support an accelerated approval pathway with confirmatory data to be provided at a later date. Another option is a traditional full approval. We are pleased to announce FDA's acceptance of our rolling BLA submission, and based on that, Capricor intends to initiate the filing of our BLA shortly."
"Should the HOPE-3 data be necessary for the BLA, we would anticipate that the submission would be complete at the end of the first quarter"
“SHOULD THE HOPE 3 DATA BE NECESSARY FOR THE BLA” → translation, we met the FDA and they said they were open to other options. The little things matter. Every change is approved by a lawyer.
A keen observer sees a change in the language. (I can't wait until ChatGPT can do this)

The Q2 Call was August 7th. On September 17, they sell the EU rights to NS pharma
On September 24, They announced plans to file for full approval with no additional data required. CAP-1002 data in 20 patients which has been available for 2 years is now enough to support full approval for DMD - Cardiomyopathy. The company will not report Cohort A data in 4Q24 (likely because it places them at risk of gaining approval).The HOPE 3 trial will serve for label expansion. In all likelihood, CAPR will have an approved therapy based on data from < 20 total treated patients in the HOPE 2 trial. The FDA CAME TO THEM with a revised position.
So it seems the following are true:
1) the majority of their trials have failed
2) they have had safety issues in the past (changed trial design to mitigate this which has worked)
3) FDA lenience has saved them from reporting a risky trial (HOPE 3). FDA seems to have come to them and asked them to apply for full approval.
4) The data shows some effect on DMD cardiomyopathy in small N
5) they should have an approved drug in DMD cardiomyopathy but have partnered away > 50% of the revenue. No data successful in any other diseases.
Capricor finds itself in an interesting position. They have a valuable asset which will likely be approved for DMD but has partnered with NS pharma and given away the majority of the revenue (50%+): Nothing much more than “Meaningful mid range doubel digit share of product revenue….between 30 and 50%.”
Side note: maybe it’s time to take a look at NS pharma (Nippon) trading at a 2B cap owning 50%+ of sales and 7% in CAPR?
One can model revenue, etc based on those above. I'm personally not one to own it. Simply because there isn't enough prior clinical data to judge HOPE-3 and it's out of my wheelhouse to trade volatile stocks.
Friedreich's Ataxia(FA) and Lexeo - Can history repeat?
Nonetheless, as investors, we can use the changing tides of the FDA to explore opportunities. Skyclarys (omaveloxolone) for Friedreich's ataxia was one of the aforementioned controversial drugs. The FDA originally didn't find the data convincing, flipped after a patient advocate petition (link). The proposed advisory committee was cancelled, and the drug was eventually approved. Skyclarys costs $300k+/year with 5000 diagnosed per year in the USA (link)
"Given the serious and life-threatening nature of FA and the substantial unmet need with no approved treatments, some level of uncertainty is acceptable in this instance and consideration of these results in the context of regulatory flexibility is appropriate." Summary review from Skyclarys’ application
The decision even comes from FDA's CDER division evaluating small molecules which is notably more stringent than then CBER division evaluating Biologics.
Lexeo

Lexeo expects to update us on their regulatory plans in Friedrich's ataxia in Q4. LX2006 is an AAV gene therapy targeting Friedrich's Ataxia cardiomyopathy, the major cause of death in FA. Patients with FA present with severe neurological symptoms but most actually die from a slowly progressing hypertrophic cardiomyopathy. The etiology is unclear: Frataxin is required to clear iron from cells, but that doesn't drive the disease. Higher Frataxin is correlated with better results but the precise level required is unclear. A few natural history studies seem to indicate multiple phenotypes of FA-CM. 80% have stable disease over time, but 20% have a rapidly progressing form of cardiac disease. (link) Life expectancy is < 45 years old.
Prior data
Lexeo presented Phase 1 interim results, but it's still very early to make a judgment. The phase 1 trial has three cohorts testing different doses. The interim results in July 2024 were from cohorts 1 and 2 while cohort 3 will report results next year.

They were supposed to report an additional biopsy in fall 2024, but they haven’t advertised where they will present the data. Potentially ESGCT October 22-25.
The data is so-so. Only two patients out to month 18. The frataxin expression doesn’t reach 5%. The company only presents data on patients with elevated LVMI. With such a small N, it’s hard to judge. Still, it’s encouraging to see functional reductions in parameters such as LVMI, troponin, and improvement in frataxin expression.
My takeaways on the data and regulatory situation
The disease progression is too slow to show any meaningful change in 12 months. Reduction in LVMI and increased Frataxin expression is the best they could show.
Enrolled patients had a normal to severe phenotype. The therapy likely benefits those with a severe phenotype the most.
More data is coming from a higher dose cohort, cohort 3 next year.
Management guided to the following targets as goals for accelerated approval with the FDA: 5% frataxin expression, 40-50% IHC, 10% reduction in LVMI, and 30% improvement in troponin.
IHC measures % of cells expressing the protein, Frataxin level measures % of normal total protein.
They're relying heavily on FDA leniency and unmet needs. I find this argument less compelling than in DMD (though still important).
Bottom Line: Lexeo will show their hand soon and It’s one way to bet on increased regulatory flexibility. The stock has fallen off a cliff recently, so I’m worried what management is saying. The aforementioned targets for frataxin expression and LVMI reduction would be a boon because lower dose cohorts have already met this bar. As they advance the drug with higher doses, results should improve. Even restricting the trial to a progressive patient population would be a billion dollar market.
Rocket's Pharmaceuticals set the precedent
Rocket has what Lexeo wants: A clear path to approval with strong data to back it up. Rocket is seeking accelerated approval for their AAV gene therapy targeting Danon disease (RP-A501). The FDA agreed to a 1-year, natural history controlled, single arm study for approval. The primary endpoint is a combination of LAMP2 protein expression (>=grade 1 IHC, 25% expressing cells) and Left Ventricular Mass Index (> 10% reduction) at 12 months with supporting secondary markers. (link)
Danon Disease Notes:
LAMP2 is a lysosomal cleanup protein where lack of expression leads to accumulation of ‘junk’ in cardiac cells. LAMP2 is a janitor.
Prevalence unknown but estimated 10000 in the USA with incidence 500/year. Danon is a relatively new disease (< 25 years old, link) but there is clear consensus about the unmet need.
Males have worse phenotype (X-Linked) and are expected to die by 20 years old
No approved therapies so diagnosis rates are unclear.
Data to date meets the bar FDA set and there is a clear causal line in my opinion from LAMP2 protein and disease manifestation. In the phase 1 trial, most treated patients have improvements in LVMI >10% and reach LAMP2 expression > 1 at 12 months (link). Most improvements are sustained over time but even with some turnover where long term expression is questionable, the primary endpoint doesn’t extend beyond 12 months. Remember Exondys is still under accelerated approval from 2016.
Enrollment in the pivotal trial completed in September so I expect data later next year. if approved, Rocket would have a gene therapy for a fatal orphan disease with no other treatment options targeting a multi billion dollar market. Patient investors could be rewarded.
On both Lexeo and Rocket, the details matter less than the overarching theme: The FDA is increasingly lenient for rare diseases.
Conclusion:
I’m a believer in regulatory flexibility as a way to increase access to medications but the process has been perverted. Flexibility in DMD was originally praised but even patient advocates are tired of false promises from drug developers. Recently, the FDA extended their blessing to Capricor by asking them to submit for full approval using iffy data; I wouldn’t bet against approval.

Agree or disagree, the FDA has changed their standard and investors should adapt. Lexeo Therapeutics is another company with an expected regulatory catalyst. Friedrich’s ataxia is another disease with high unmet need (though less than DMD) so I think FDA lenience is plausible (we’ve already seen it with Skyclarys). One precedent in the cardiomyopathy space is Rocket Pharmaceuticals who is developing a gene therapy for Danon Disease. The FDA set a low bar for their accelerated approval.
Would love to hear y’all’s thoughts: do think regulatory flexibility is good? Do you think FDA loves DMD too much? What are some other examples? Do you disagree with any of my interpretation?
Adu
Not investment advice: I may hold positions in the securities mentioned.
Defining terms
Left Ventricular Mass Index (LVMI): LVMI is a measurement that shows the size of the heart's main pumping chamber (the left ventricle) relative to a person's body size.
Immunohistochemistry (IHC): measures total percentage of cells expressing a protein
Total protein expression: Overall amount of a particular protein that cells or tissues are producing.
Troponin: protein found in heart muscle that gets released into the blood when the heart is damaged
BLA acceptance or refusal next week! It was a rolling BLA with the first section submitted prior to the other 2. I fully anticipate it being accepted. They've met with the FDA >20 times. Institutional ownership has skyrocket and continues to tick upward everytime a new report is published.