
microRNA Wins a Nobel: why no successful drugs?
Quick history of miRNA science + a look at RGLS and ADPKD (regulatory flexibility shows up again)

There are a few posts which satisfy nothing but my intellectual curiosity. This is one of them.
This year’s Nobel Prize in Medicine, awarded “for the discovery of microRNA,” was a reminder of the promise that once surrounded microRNA (miRNA) therapeutics. Numerous drug candidates were developed in the mid-2010s, yet each failed—whether due to limited efficacy, toxicity issues, or financial constraints. Multiple companies pivoted: Miragen rebranded as Viridian, MiRNA Therapeutics transformed into Synlogic, and the only public miRNA company remaining is Regulus Therapeutics. (Novo Nordisk did acquire Cardiol for $1B last year, but the field is notably thin.). If you’re a fan of the Incredibles, I felt like Mr. Incredible going through the history.
Why haven’t miRNA drugs cracked the code? What is the "rate-limiting step" holding this modality back? And does Regulus stand a chance?
A Glance at miRNA Science: “Something Weird in Worms”
Originally thought to be “something weird in worms”, microRNA regulation was discovered in 1993 but forgotten until the early 2000s when we discovered conserved miRNA sequences in multiple animal species. A number of researchers then showed how miRNAs are implicated in biology and the role they play in normal development and disease.
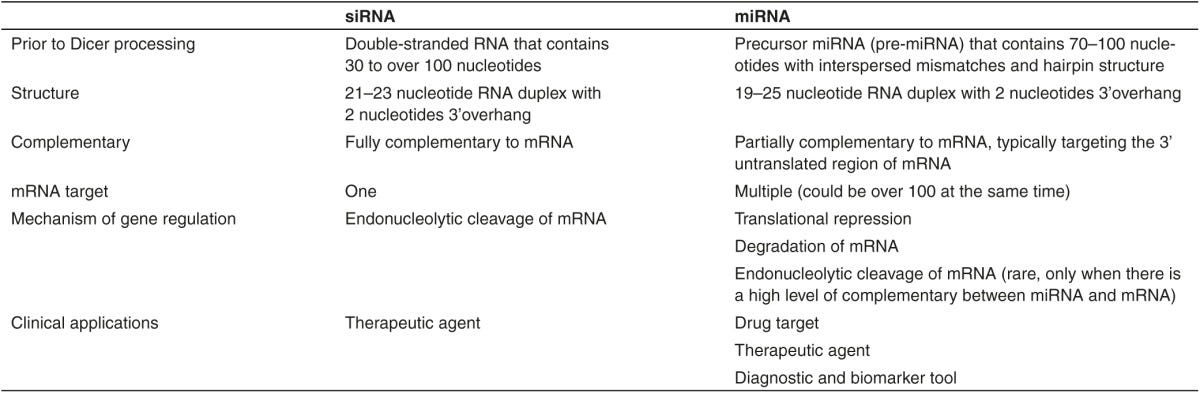
Let's lay out the steps to make a protein: DNA -> transcribed into mRNA -> Post translational modification -> Translated into proteins. microRNA is somewhat similar to siRNA (small interfering RNA, what forms the basis of RNA interference). A microRNA regulates formation of proteins at the mRNA level by using silencing complexes to bind mRNA transcripts and break them down. siRNA is highly specific for one mRNA while miRNA uses a 7-8 base pair long ‘seed sequence to bind different mRNA (source).
A miRNA is transcribed in the nucleus from DNA, undergo some cellular processing to cleave them into functional molecules and act in the cytoplasm through multiple, context dependent, methods. The pathway for creation can vary, but the details are beyond the scope of what’s necessary to understand how they work at a high level.
Nature of regulation
 - Wikipedia")
microRNA is implicated in multiple species for normal development and disease processes, but the regulation is not straightforward. They are both promiscuous and have a shallow impact on protein levels.
The most important principle: microRNAs are a promiscuous regulator. One miRNA affects hundreds of genes to create complex patterns of gene expression. Think of miRNAs like a sculptor placing the final touches on an already molded piece of marble. miRNA is a key regulator in both disease and normal development, but they don’t draw a 1-1 line from molecule to effect.
An additional key insight is that miRNA acts at the RNA level and not the protein level which has two key implications: 1) measuring mRNA levels is a good proxy for miRNA activity (good for early drug developer using biomarkers) and 2) the impact of miRNA on protein can be marginal (terrible because we need significant changes for therapeutic efficacy)
We can see the effects of miRNA in a landmark paper from 2010 testing the effects of miR-223 in humans and protein expression. Researchers investigated loss of miR-223 and protein/mRNA changes and concluded “for most interactions microRNAs act as rheostats to make fine-scale adjustments to protein output”. (link) We want to see > 10-fold differences for therapeutic efficacy (siRNA shows 80%+ silencing, link). miRNAs barely affect protein levels.

This chart looks like noise, but it speaks volumes. Three key takeaways from the graph
miR 223 affects a shit ton of genes, Look at the number of dots on that graph!
The correlation between mRNA and protein expression is nearly 1:1 indicating regulation of protein expression likely occurs at the mRNA level. No additional silencing occurs
miRNA impact on protein levels Is modest. Loss of miR-223 leads to a maximum 2-3 fold change in protein levels. Not Great.
This dual challenge—miRNA's broad target range and its marginal effect on protein levels—creates hurdles for therapeutic development.
Rate Limiting Step: Why hasn’t miRNA been successful
The roadblocks for miRNA drug development reflect the complexity inherent in biology. An ideal therapeutic pathway has clear pharmacology, well-defined targets, reliable preclinical models, and straightforward clinical trial endpoints. There should be a straight line from drug to benefit. But miRNA therapies lack these advantages. In addition to the traditional delivery challenges for nucleotide-based therapies, the drug and disease biology is unclear. Further, the promiscuity leads to off target effects which are unpredictable and a broad but small impact on protein levels leads to high levels of noise. We can’t tune miRNA to our desires unlike siRNA because the rate limiting step for miRNA is partially inherent to our biology.
Contrast those issues with the development of RNA interference. RNAi was left for dead after a flurry of interest in the early 2010s (it won the 2006 nobel prize!). Roche, Pfizer, and Merck all shut down their research centers and Novartis dumped the platform which became Alnylam. Nothing had been successful, but the science was clearer cut: if you can deliver the molecule effectively, the drug should work.
“The major bottleneck in the development of RNAi therapies is the delivery of these macromolecules to the target cells.”
And Alnylam solved the delivery issue by discovering GalNAC conjugates. The story is clearly not a straight line, but I think it demonstrates an important principle: effective drugs often solve tractable problems: History here.
The same principles I mentioned in my October 2024 Radiopharma Article apply:
“Dosimetry turns biology into physics. IO is a minefield of early stage results not translating in larger trials because it's a biology problem. Dosimetry is a physics problem: we need to deliver a dose to the tumor to kill it. Can we get that dose without off-target toxicity? Early stage, in-human dosimetry translates well into late stage results for efficacy (with small N ~10-20). Comparable patient populations should show comparable dosimetry results and dose = response.”
I believed radiopharmaceutical therapeutics had solvable physics problems rather than intractable biology problems which give them inherent advantages. The thesis has yet to fully play out, but my thought process applies.
A case study of failure: RG-101 (miR-122)
RG-101 was a once-promising drug candidate that failed in phase 2 trial because of unexpected side effects. The phase 1 data was great, but the Phase 2 trial failed due to toxicity issues—specifically, hyperbilirubinemia. How did this happen? None of its main target genes were linked to hyperbilirubinemia, but when researchers looked closer, a few of the *70+* off-target genes showed connections to jaundice. Yes, that’s correct: Seventy plus off target genes.
And there lies the core issue: “Most miRNAs target tens, if not hundreds, of genes, a phenomenon [researchers] call ‘Too Many Targets for miRNA Effect’ (TMTME).” You can’t hit on everyone in the bar if you want to get someone’s number. (source)

miRNA has major issues, but can Regulus thread the needle?
Regulus and ADPKD
Amid this backdrop, Regulus Therapeutics presents an interesting case with its miRNA drug candidate RGLS-8429, designed to treat Autosomal Dominant Polycystic Kidney Disease (ADPKD). Regulus may have a unique opportunity within ADPKD’s specific pathology and FDA leniency even with a frustrating history of failure. I will caution investors that A) Regulus is an extremely tiny company b) their drugs have failed in the past and c) they effectively have on drug in development. In sum, this is not a stock pitch. I simply found the drug and science interesting.
Background on ADPKD:
(ADPKD) is a genetic disorder causing numerous fluid-filled cysts to develop in the kidneys often leading to kidney failure requiring transplantation. In fact, ADPKD is one of the leading causes of kidney failure in the USA. The only approved treatment, Tolvaptan, is a vasopressin antagonist, originally approved in a different indication. Sold as Jynarque for ADPKD, the drug, slows eGFR decline (measure of kidney function), but comes with a black box warning for liver toxicity. The unmet need for a fatal disease with no disease modifying therapy is high. Tolvaptan barely slows disease progression yet still annualizes nearly $1B in sales (source, source)
ADPKD is caused by a mutation in the PKD1 (80% of cases) or PKD2 gene (15% of cases) leading to lower polycystin levels. Lower Polycystin leads to cyst formation. With evidence pointing to a clear correlation between ADPKD disease course and genetics “Hypomorphic PKD1 alleles can cause mild kidney disease or liver cysts in the absence of clinically manifest kidney involvement. The presented data highlight pleiotropic ADPKD clinical presentations and varying severity of kidney disease from PKD1 allele combinations.“ (source). Patients can progress more slowly if the mutated PKD1 gene preserves some activity.
The predominant theory for pathogenesis is a “two hit hypothesis”. Cells with an underlying mutation in PKD1/2 acquire another mutation which reduces Polycystin levels below a threshold leading to cystic growth.
“Recent evidence indicates that significant reduction of functional PC1 expression below a critical threshold level is sufficient to result in cyst formation in some situations” (source)
Further, it seems “subtle differences in PC1 levels have a dramatic effect on the rate of cyst formation and/or growth.” (source)
The evidence is strong that polycystin levels are correlated with disease progression and restoration of polycystin reverses the disease course in animal models. (source). Increasing Polycystin by 1.5 fold compared to controls effectively reversed the disease in mice.
Conclusion: both preclinical and human genetic evidence support polycystic as a key regulator which can slow and potentially reverse disease progression. The role of miR-17 in ADPKD is less clear and the same “broad but shallow” effects are hard to parse (source)
Side note: defining TKV, Mayo classification
Height adjusted Total Kidney volume is a potential prognostic tool for ADPKD disease progression. By measuring TKV and adjusting it for height, the mayo imaging classification (MIC) sorts patients into one of five classes (1A to 1E), each reflecting a different TKV growth rate, from low risk (<1.5% annual growth) to high risk (>6% annual growth). Usually patients in class 1C -1E will show increases in TKV year over year.
Could the drug actually work?
RGLS-8429 is a second generation drug targeting miR-1. The proposed mechanism of action is two fold: 1) increase polycystin by blocking miR-17 and 2) a vague notion of miR-17 dysregulation in ADPKD. The first generation molecule, RGLS 4326, showed efficacy in preclinical models and modest efficacy in patients but was discontinued due to off-target CNS toxicity (Development). The new drug, 8429 avoids CNS toxicity and shows preferential accumulation in kidney cyst cells (solves some of the delivery issue).
RGLS-8429 is in a multiple ascending dose study (MAD) testing effects on kidney volume (measures cysts in the kidney) and polycystin levels. The results are in the latest investor presentation and they are….mixed:
My TLDR
Polycystin change doesn’t meet the bar: Polycystin change is < 50% for both PC1 and PC2 which doesn’t meet the preclinical efficacy bar (1.5 fold increase). Management proposes mir-17 is more broadly implicated in disease pathogenesis, much less evidence for this
Noise: The polycystin results are all over the place, and eGFR (kidney function) didn’t improve, which means it could take years to see any real effect. (Regulus is hoping the FDA will accept total kidney volume, or TKV, as a surrogate endpoint for accelerated approval.)

Early signs in TKV: The drug shows early slowing and potential reversal of TKV growth. As mentioned before, Class 1C -1E patients shouldn’t improve with respect to TKV growth.

Dose response unclear: Small N, variable baseline eGFR, individual patient data is all over the place

Regulatory flexibility again:


But none of the mess of a dataset matters if the FDA gives them the go ahead for accelerated approval using height adjusted kidney volume and they show some benefit. The end of phase 1 meeting is expected by the end of 2024 and Regulus wants accelerated approval based on TKV.
Tolvaptan review vs now
“Today’s FDA is not Yesterday’s FDA”. The FDA scoffed at using TKV as
a surrogate marker for approval, requiring Otsuka to run a multi-year study to show functional benefit. This was the FDA’s perspective on Tolvaptan:
“the Agency has consistently denied effects on kidney volume as an adequate surrogate for progressive loss of renal function” - FDA review document
Contrast that perspective with now:
“The FDA designated TKV as a reasonably likely surrogate marker for disease progression in ADPKD, which could serve as an endpoint under an accelerated approval pathway” - 2023 summit

So here we are once again. A drug shows some benefit on a surrogate endpoint for a disease with unmet need: sound familiar? Science be damned. FDA lenience has proven forgiving for multiple drug developers starting with SRPT, moving to BIIB, CAPR, RETA, etc. Regulus could follow. A phase 3 trial evaluating eGFR would require multiple years of followup but a 12 month TKV trial is much shorter.
Regulus expects an end of phase 1 meeting by the end of the year and will report more results in Q1 2025 from the MAD study. A positive outcome would be improvements in TKV and confirmation from the FDA that TKV is a worthy surrogate outcome.
As mentioned before, Regulus is a one product company with messy science and prior failures. However, Regulus could thread the needle for an effective miRNA therapeutic for the following reasons
ADPKD has a high unmet need so the FDA may accept marginal efficacy
miR-17 is implicated in ADPKD -> silencing it can improve the multifactorial disease
RGLS8429 preferentially targets cyst cells -> partial solution to delivery issues
Genetic evidence shows a dose dependent effect of polycystin levels on ADPKD progression -> maybe small increases are adequate
It’s an intriguing situation which I’ll be tracking as it progresses.
Conclusion
The discrepancy between good science and good drugs teaches us how difficult drug development can be. Nobel prizes tend to precede period of heavy investment (RNAi in 2006) or follow development of revolutionary therapies (Immunotherapy), but miRNA fits a unique spot: we’ve already tried, unsuccessfully, multiple drugs. The problems using miRNA therapy seem intractable so it’s hard to imagine a near term revolution. However, a combination of increased regulatory flexibility and the specific qualities of RGLS8429 pique my interest