


T Cell Engagers in Auto Immune disease
Why they should work and the one public company with a drug in development

TLDR
CAR-T shows durable remission in Auto immune disease
T Cell engagers are starting to match CAR-T in Oncology efficacy
T Cell engagers can match CAR-T efficacy in Auto immune disease
The disease burden is less (less B cells)
Next generation drugs are better than TCEs currently cited as failed proof of concept in auto immune disease (CD20 lunsumio, CD19 Blincyto, CD19 A-319)
The focus is on defining the parameters for an immune reset and why CD19 T cell engagers can induce the reset needed. I originally wanted to pitch CGEM as a stock but after recent management commentary, I think the Q4 Readout is hard to get confident. Still, T cell engagers will work

The core question underpinning the article: Can we give a safe and commercially viable one time therapy to severe autoimmune disease (AID) patients to cure them of disease? Yes
Summary of the article and key questions answered
Autoimmune disease: Our cells attack our cells. Driven by B cells, T cells, and cytokines. Often multifactorial, but specific pathogenic mechanisms are implicated in each disease.
What is an Immune reset? We wipe out the immune system and a patient with auto immune disease no longer shows clinical activity even after their immune system repopulates. The new B/T cells aren’t pathogenic
Similarities to Lymphoma? Lymphoma is a B cell disease (i.e. too many B cells). Drug development there gives us information on how the progression from CAR-T to T cell engagers and other modalities may progress. Furthermore, efficacy in Oncology likely correlates to strength of depletion in Auto immune disease. WE can also use specific subtypes (Follicular lymphoma vs DLBCL vs B-ALL) to understand what drives efficacy
Differences? Lymphoma has a higher B cell burden and thus requires higher doses of CAR-T, lymphodepletion, etc. Oncology patients are willing to tolerate more toxicity.
Lymphodepletion: We use Lymphodepletion to prep patients for CAR-T by killing their immune system. Some say it contributes to the efficacy (we’re killing B cells after all) but I don’t think that’s true in auto immune disease.1
What drives the toxicity (i.e. CRS) and efficacy of T Cell engagers? Efficacy = potency X AUC concentration. Toxicity = CMax.
How does B-cell depletion affect the immune system and its ability to fight infections? Long term depletion can lead to infections, but the goal is repopulated the immune system after a reset.
Does the specific disease matter? Yes. Killing all the B cells won’t work if the disease is mediated by T cells. Lupus is a good disease becasue it’s driven by B cells but hard to treat. Other diseases like Myasthenia Gravis are driven by antibodies so a “reset” isn’t needed for highly effective diseases.
Near term Catalysts and important information to track: I’m tracking any upcoming auto immune conference, updates from Cullinan on CLN-978, Merck on CN-201, AZN on Blinatumomab and Uplinza
The Immune Reset: CAR-T Works
CAR-T works: George Schett’s landmark paper using Car-T to treat refractory autoimmune diseases shows 92% remission rates out to 5 years. Patients were cured after a one time treatment
But how the hell did we have the idea to take a toxic, next generation oncology modality and target autoimmune disease to create an “immune reset”?
The idea for an immune reset started in the 1980s with Hematopoetic Stem Cell transplantation (HSCT). Back in ye olde day, before iphones, ChatGPT, and twitter (Yes, to some, I’m dating myself as a young person), we used HSCT to treat severe autoimmune diseases.
Note: For basics on auto immune disease: see this deep research report, Or use ChatGPT to ask questions as they come up. I will not rehash them here.
HSCT works through an immune reset
Hematopoetic Stem Cell therapy uses Lymphoablation to wipe out our native immune system (hyperactive in auto immune disease) and “reset it” by giving back naive stem cells. It was first proposed in 1995 and with the first clinical case report in 1996. Physicians saw it work to induce "remission" and they came togeher study the outcomes. Over 15 years, ~1000 patients were treated with HSCT, primarily in Multiple sclerosis, Systemic sclerosis, and Lupus. The original trials show ~40% of patients are disease free out to 5 years with an overall survival rate of 80% and high between center variance in outcomes.
The HSCT uses the following to induce a “reset”
Kill all the immune cells in your body
Give your body new stem cells
Hope it works
But it has major problems. HSCT is an art reserved for the physicians at specialized academic centers, not a commercially viable treatment. Yet it still gives us a But this gave us proof of concept. Yes, people died from the transplant. Yes, only 40% of patients were in 5 year remission, yes it’s highly center dependent. But the HSCT shows us an immune reset is possible. We can give back stem cells and induce remission for severe disease. It's even covered by insurance for severe systemic sclerosis.
Enter CAR-T
CAR-T for auto immune disease is trying to do the same thing as HSCT but in a more targeted approach. Instead of complete lymphoablation, we use Lymphodepletion to reduce the burden of immune cells and then we give a CAR-T to target and kill all cells expressing a target (CD19 was the original target).

CAR T-cell therapy in autoimmune diseases - The Lancet
Stepping from HSCT to CAR-T is analogous to the development of Lymphoma therapies. Standard upfront treatment is R-CHOP and stem cell transplant after failure. CAR-T is often used in the 3rd like setting and progressively moving earlier. We’ll discuss analogies to oncology later.
For autoimmune disease, instead of destroying the whole immune system with HSCT, CAR-T targets specific pathogenic clones. In theory, we only need to “reset” CD19 positive cells. Note CD19 positive cells are the earliest stage of development. However, they’re not expressed on mature B cells or T cells so a CD19 CAR-T doesn’t touch that. In cancer, this isn’t an issue because the disease is b cell mediated.
But for AID, targeting CD19 doesn’t sound right: AID are often multifactorial with a T cell component. Some are B cell mediated but for most hard to treat conditions, the interplay between T cells and B cells is unclear. Can you still effectively clear all the B cells in the body enough to induce a reset with a CAR-T? Naturally, 2 separate questions have to be asked.
If we can reset the B cell compartment targeting only CD19+ cells, is that enough to induce sustained response in certain AIDs?
Can CAR-T match lymphoablation? (Do we even need to)?
An academic will hedge: I won’t.
The answer to both questions is yes.
The recent George Schett data proves the biology works: we can induce an immune reset in Lupus leading to a clinical remission.
In Oncology, CAR-T B cell depletion matches ablation and transplant
But the proof of concept, pioneering therapy has issues.
Issues with Autologous Car-T:
Issue 1: Small Patient population with refractory disease willing to undergo treatment
Autologous Car-T for Auto immune disease is an exercise in commercial futility. The academic centers should use CAR-T to demonstrate proof of concept, but the manufacturing constraints, lymphodepletion required, and patient population make traditional CAR-T unfeasible. One eligibility study used an 1800 SLE patient cohort and showed only 54 were eligible for treatment and only 2 ended up getting it. 2 people. Only 1/1000 were eligible based on the severity of disease.
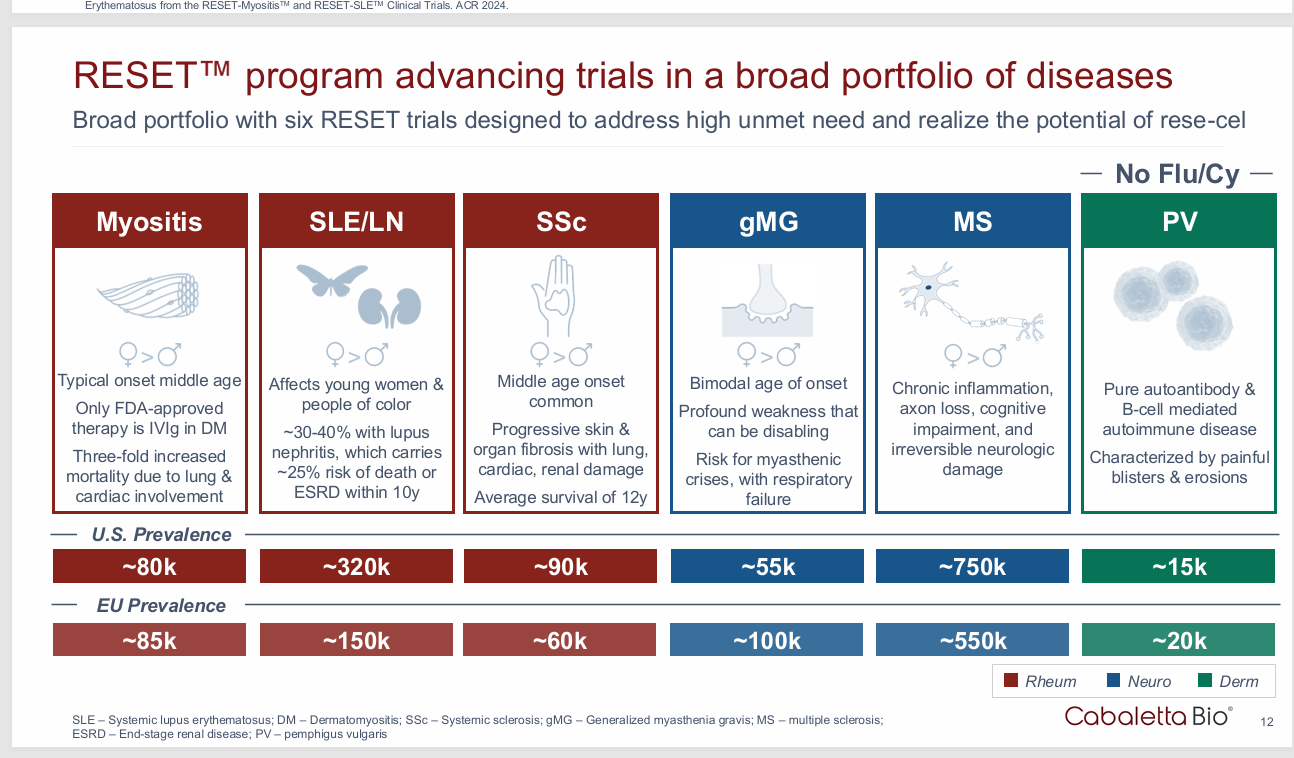
This should be 500 eligible.
Patients were required to have extensive disease, active disease, the right manifestation and even when a patient meets all the criteria, they can’t stay in the hospital for that long.
I met the criteria for the study;however, it was wayyy more extreme than I thought it’d be so I didn’t go through with it.
Required a week long hospital stay where they basically shut down your entire immune system and then gave you the CART therapy and then it required someone taking care of you for a while because you would be so weak and barely able to walk and frequent doctors appts. I couldn’t commit to all that. I’m already not working, we can’t afford for my husband to also not work to take care of me
This isn’t an aggressive B cell cancer with a 5 year life expectancy: patients live *somewhat* regular lives, but they know sometime down the road, their cocktail of medications, kidney damage and chronic pain will kill them.
Issue 2: Costs and Manufacturing
We can’t make enough doses. Cabaletta bio is scaling to 1000 doses per year in 2027 (and we all know that’s a projection, not a promise) and they’re the leading company. Administration for an Autologous CAR-T requires a weeks long process in the hospital with lymphodepletion. This cannot be scaled. Refractory auto immune patients don’t havce a direct pipeline to large centers like cancer patients. They just deal with their symptoms for life.
Question: Does the unmet need even exist?
Auto immune disease won’t kill you…fast. It kills you slowly, sapping the life force via side effects from flares, chronic immunosuppression. Lupus patients won’t die in a few years, but they eventually require dialysis for kidney failure.
Question: Can we avoid Lymphodepletion?
Lymphodepletion suppresses the immune system to create space for the CAR-T cells. The conditioning kills existing cells to reduce competition for the new CAR-T cells and creates a favorable environment via the right cytokines. In oncology, higher intensity depletion often leads to a favorable cytokine profile associated with longer progression free survival (PFS) and better results. But this is some once daily pill: it’s a month-long hospital course turning you into a”bubby boy” with no immune system. If we don’t need, we shouldn’t use it

There’s an interesting conundrum: if the goal is to kill our immune cells and the lymphodepletion is depleting our immune system, does it serve a bigger role than just “conditioning”? Some theorize it contributes to efficacy too.
The theory is supported by data in oncology. CHOP is a pseudo depleting first line lymphoma therapy and isn’t fully immune ablative yet still induces remissions. And high dose Lymphodepletion can often lead to better results. But the data in auto immune disease is different.
I don’t think heavy lymphodepletion contributes to efficacy for a few reasons.
We’ve tried it: Lymphodepletion alone doesn’t induce clinical responses in AID. We’ve tried LD to induce immune resets and it just doesn’t work. Most recently, George Schett treated a relapsed CAR-T patient with Lymphodepletion with no clinical response.
“Despite full-dose lymphodepletion, no clinical response was observed. (in auto immune disease)”
Lower burden of disease: LD works in Oncology in part by increasing the concentration and persistence of CAR-T Cells. We want high initial peaks and long term CAR-T persistence in cancer. But the Schett data actually shows us the opposite in autoimmune disease. After an initial spike in CAR-T cells, concentrations drop to 0 AND we still see continued responses. We don’t need CAR-T cells around forever.

We’ve seen proof of concept from FATE Therapeutics treating patients without lymphodepletion. They show n=1 data *requires a pillar of salt but it demonstrates some proof of concept. Unfortunately, the B cell depletion wasn’t complete which means higher dosing or potent drugs are required induce durable clinical responses.
Question: Why target CD19, not CD20?
CD20 was the original target for immune depletion. Rituximab, a CD20 targeting antibody is used to treat lupus to reduce pathogenic B cells, yet some patients still relapse because CD20 is expressed on a narrower subset of cells. Patients who respond to rituximab and then relapse often have CD20 negative, CD19 positive B cells still floating around. Plus, CD19 CAR-T can work AFTER rituximab. Furthermore, recent data from mosunetuzumab in Lupus (CD20 T Cell Engager) is worse than all the CD19 T cell engager data even though mosunetuzumab works better in Lymphoma. We’ve seen CD20 data disappoint repeatedly and this isn’t a drug issue: it’s a target issue. All proof of concept data supports CD19 as a target. If we’re resetting the system: why not go after the earliest cells in development?

Question: Depletion vs Clinical Efficacy
I use B Cell Depletion as a proxy for efficacy becuase clinical responses (i.e. how patients feel) can be swayed by placebo. Clinical responses are the goal and inducing durable long term remission isn’t fake, but early phase clinical data is hard to interpret. In my opinion, B cell depletion correlated to clinical response is a good proxy
Next up: T cell Engagers and lessons From Oncology
In Oncology, the next step after CAR-T (Autologous or Allogeneic, T cell or NK cell, lymphodepletion vs not) is a T cell engager.2 Remember the goal is B cell depletion and the method doesn’t matter.

A CAR-T therapy infuses outside T cells to kill but a T cell engager (bispecific T cell engager, BiTE) uses the patient’s T cells to do the killing. The TCE has a target on one end and a CD3 moiety on the other end. The target engages the target cell and recruits T cells to kill them.
CAR-T = T Cells with target on surface. TCE = Recruit T cells

T cell engagers are given IV (usually) in the hospital/clinic to watch patients for “cytokine release syndrome” (CRS) where activation of the immune system causes fevers, chills, etc or Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS). They’re dosed for a fixed number of cycles until either remission or progression of disease. They are not forever therapies. T Cell engagers can be given for a set amount of time.
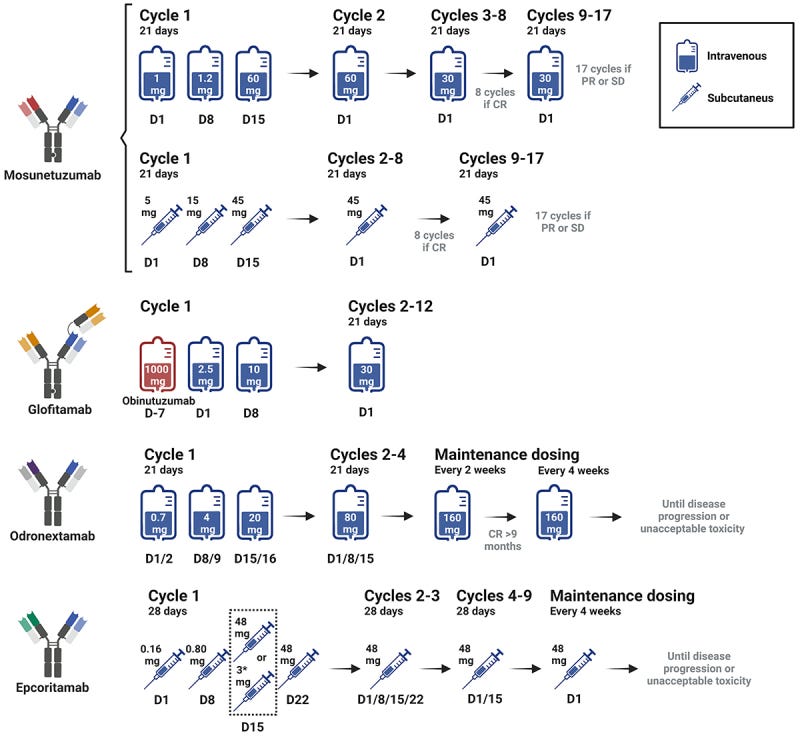
Lunsomio dosing for 8 cycles induces 70% CR rates in FL. A sample of Four TCEs are approved across multiple lymphoma indications3.

But one Lymphoma != all lymphomas. Some have a higher burden of disease, a higher proliferation or distribution in blood compared to lymphoid tissues. Tissue Lymphomas with high proliferation rates are harder to treat than blood based, indolent disease so CAR-T (often more effective, more risky) is preferred in a disease like DLBCL. T cells are readily available in our blood for killing while tissue based disease is harder to eradicate. Furthermore, without enough T cells, they can become “exhausted”, essentially quitting because we’re working them too hard like a Gen Z employee

But the comparative efficacy data is not a modality limitation, it’s a drug design problem. The right TCE can match or exceed CAR-T efficacy.
I recommend the following for a good review comparing BsABs (TCEs) and CAR-T in Non Hodgkin’s Lymphoma. https://www.mdpi.com/2072-6694/17/7/1192. The paper details recent trials well.
We can use years of experience in Oncology to guide our reasoning. Remember the goal is B cell depletion. Better depletion, specifically in tissues, leads to more efficacy.

Blinatumamab efficacy lessons - We can design Better TCEs
Blinatumomab is approved for ALL but has been studied (unsuccessfully) in NHL.
This paper analyzed the PK and PD for blinatumomab in NHL.
Dose is linearly related to the Steady state concentration
")
But the B cell depletion is related to the steady state concentration in a logarithmic fashion.
")
The paper tells us the minimum dose required is 47 ug/m2/day, nearly double the FDA approved dose for cancer. Blinatumomab could work in NHL, but the efficacy is limited by the tolerability. We tried dosing up to 112 ug!, but too many people dropped out.
Non hodgkin’s lymphoma (NHL) is also a tissue based disease which requires deep tissue depletion yet Blinatuomomab doesn’t penetrate tissues Peripheral B cell depletion is easy but the kinetics inform us max efficacy was never reached. . Using the dose response curve, we can reach 10000 pg/mL and still increase efficacy.
Blinatumomab is a bad drug: it’s a pioneering therapy, approved years before most other TCEs, but it has a host of issues.
It’s dosed daily with a 5 hour half life.
The drug doesn’t penetrate tissues well (hence approval only in ALL)
It lacks the Fc effector function. It’s a CD3-CD19 which doesn’t activate antibody related functions limiting efficacy.
Better CD19 bispecifics are possible

Astrazeneca’s TNB-046 shows an 80% response rate in 5 CAR-T naive patients and 36% response rates in patients who received CAR-T. It’s like a pistol doing more damage AFTER a bazooka.
From that data, we come to one major conclusion: Blinatumomab is not the best T Cell engager. In both Oncology and auto immune disease, we can design better drugs.

Oncology is not Auto immune disease.
WE can’t directly apply the efficacy lessons from Oncology into autoimmune disease. “Durability sucks, this data is nowhere near CAR-T data”, the B cells are replenished after depleting with a TCE. BUT AUTOIMMUNE DISEASE IS DIFFERENT. B cell lymphoma is driven by B cells that are DESIGNED to proliferate. Cancer is, at its core, a disease of proliferation.
Auto immune disease is different.
The B cell burden is much lower so we need less killing.
Dosing so far hasn’t reached therapeutics levels in Onc˜ology
Short half life drugs limit the potential efficacy due to safety concerns.
Lack of tissue penetration aka Blinatumomab is a bad drug
We don’t need a bazooka like CAR-T, it’s okay to treat it with
What about safety with T Cell Engagers? CRS Risk?
T cell Engagers are much safer than CAR-T but we’re still concerned about 1) CRS/ICANS related to the drug itself and 2) infections from depleting our B cells. Cytokine release syndrome is an immune mediated side effect where t cell activation leads to excess cytokine release immediately and causing immune related symptoms like fevers, chills, etc. CRS is most common after the first dose and driven by the relative affinity for CD3 and CD19. CD3 is expressed on T cells so higher affinity for CD3 leads to more activation and thus higher rates of CRS.
“Engineering a high-affinity tumour arm (CD19) paired with a low-affinity CD3 arm creates a kinetic hierarchy—“tag the target first, call the T cell second”—that maintains tumour killing but tempers cytokine peaks.” - This is ChatGPT and I tend to agree (although an oversimplification).
But we see how it played out with macrogenics’ MGD011. Their relative affinity for CD3 is 10x lower than CD19

MGD017 was discontinued for risk of CRS and safety.
The second piece to CRS is the initial concentrations in the body: CRS occurs after the first infusion driven by the Cmax (max concentration). That’s why doctors step up doses in the real world: they start slowly to avoid the safety risks because that first dose can spike cytokines.
To alleviate the CRS risk, we should want to characteristics
High relative CD19 to CD3 affinity ratio
And low initial concentration spikes.
ICANS (immune cell associated neuro toxicity syndrome) is a poorly understood mechanism of toxicity. Honestly, we can postulate on ICANS risk but I think the human data is a better guide for any potential safety issues.
The long term Safety Risk: People think we’re suppressing the immune system and cause infections
This isn’t a concern in my opinion
Patients eligible for these therapies are already on Chronic steroids or other immunosuppressants for life.
CD19 isn’t expressed on mature plasma cells so patients maintain vaccine titers
Resetting the immune system leads to repopulation of B cells
The data in Auto immune disease
CD19 TCE data
T Cell Engagers were used to treat severe RA patients, Systemic Sclerosis, and Lupus patients, Anti-synthetase syndrome. (most use blinatuomab, another uses A-319).

In each trial, they underdose compared to the oncology data and still induce a remission. People will pick apart this data becuase the patients relapse, the depletion relapses, etc.
They’re wrong. We’re taking ineffective cancer drugs, under dosing them, and applying them to highly refractory auto immune disease patients. This isn’t good data. But we still see inklings of efficacy. We see clinical responses and B cell depletion. Tissue depletion in muscle biopsies (though no data on lymphoid tissue) is strong
Bull and Bear Case for Cullinan - A next generation TCE
I recommend reading this thread on twitter from Adam May, Smart guy who characterizes the bull case for Cullinan well : https://x.com/A_May_MD/status/1854639638880657838
Efficacy: They engineered the drug to be high CD19 affinity and induce responses in tissues. But the Oncology data is meh, n=3 data with only one response. They’re able to treat tissue disease in a Mantle Cell lymphoma patient but B cell depletion is less than ideal.



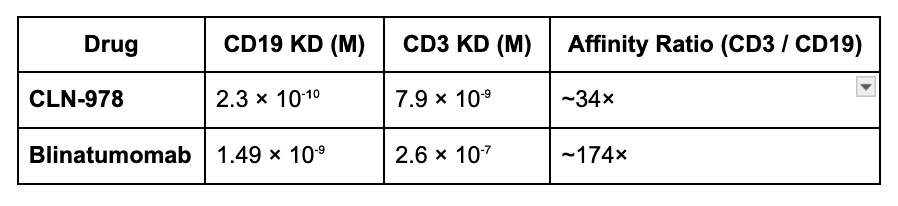
Safety: CD3: CD19 affinity is relatively high and subcutaneous dosing avoids the initial drug spike which allows for higher dosing
Compared to Blinatumomab which has a relative affinity of 1.49 nm for CD19 and 260nM for CD30, a ratio of 174 indicating the risk of CRS is much lower. CLN0978 has a relative affinity difference lower at 34 fold but a much higher CD19 affinity.
Scarcity and Why it’s a valuable asset
Merk Acquired CN201, a CD19xCD3 T cell engager for $600M even though the phase 1 data in lymphoma is approximately in line with blinatumomab. Further, it’s IV administered leading to higher rates of CRS and adding patient burden. Cullinan is currently valued at 0$ EV.
But I can’t, in good faith, pitch the stock. Yes, TCEs will work. Yes, it’s a huuge market, but recent commentary from management is negative: seems like they’re emphasizing efficacy worse than CAR-T

But let’s say they show us good data: what then? We have to run a long phase 2/3 study to get approved.
It’s certainly a valuable asset, but knowing a modality works and making money in an investment are different propositions.
Summary
CD19 CAR-T Induces immune resets with patients in clincial remission after CAR-T cell s disappear and B cell repopulate.
The natural next step is a CD19 TCE But blinatumomab is IV administered, doesn’t penetrate tissues, and is limited by toxicity.
Other CD19 TCEs in development are more effective in Cancer proving we can design better drugs.
Yet blinatumomab still demonstrates efficacy in Autoimmune disease (while still underdosed)
Supporting the theory the burden of B cells in AID is lower.
So a drug like CLN-978 which is higher potency, subcutaneously administered and some preliminary evidence of efficacy in Lymphoma could induce a reset in auto immune disease. (although the timelines are long
People don’t believe T cell Engagers can induce an immune reset. April/BAFF inhibitors, also targeting B cells, are worth multiple billions, but B cell depletion with CAR-T is worth ~0 at the moment. Furthermore, every Key opinion leader doesn’t believe in T Cell engagers because all the data we’ve seen so far are bad drugs in Auto immune disease. When we get a good CD19 T cell engager in AID, the results will be great.
Appendix: helpful sources, threads, etc.
Adam May with the Bull case for CGEM
Dimension cap on B Cell depletion
At this point, it is fairly certain high dose LD is better in oncology.
I am choosing to skip over in vivo CAR-T, allogeneic CAR-T because they haven’t worked so far. Both are great in principle and less required depletion may mean it works better. I’ll wait for more data to make a judgement
This is the list of CD19 and CD20 T cells
Great breakdown on TCEs and CAR-T for AID.
I've worked in CAR-T/CellTx for many years. If I could all of a sudden snap my fingers and remove the logistical challenges, reduce COGS, minimize residual hospital costs, make it all outpatient, make it so that it can be administered in community hospitals, scale mfg to easily meet demand, and price it competitively with antibody based drugs, I would. Turns out that that isn't possible right now, lol. It's going to be an uphill battle for cell therapy companies in this TA, especially the auto-CAR-T companies.
Insurers are going to make it tough to secure reimbursement and also put them so low on the formulary in order to manage costs. In a world where both CD19xCD3 and auto CD19 CAR-T are approved. You're going to have a way easier time getting CD19xCD3 commercially, even if it ends up falling short of the "one-and-done" bar CAR-T is shooting for.
Great read! A few things:
1. My take on management’s comments isn’t as pessimistic… they definitely highlight some nuance. It’s too early to tell anyway. Even if CAR-T ends up yielding the best results, the process required for treatment will be a massive barrier to access and utilization (leukapheresis, lymphodepletion, cell processing, etc). We are already seeing this in MM, where patient preference is bispecifics > CAR-T.
2. Was $IGMS’ invotamab’s failure due to its CD20 target or due its IgM modality? Or due to some issue with CD3 engagement? It’s hard to tell.
3. A private company, Candid Therapeutics, is advancing its CD20 TCE (CND261) and a BCMA TCE (cizutamig) into a Ph1 with RA, SLE, SE, MG and IgAN cohorts. So the story of CD20 TCEs may continue to involve. Interestingly, CGEM picked up a BCMA TCEs after this news.